
 Sample Submission Guidelines
Sample Submission Guidelines
Sanger sequencing, a classic DNA sequencing technology, has played an irreplaceable role in molecular biology research, clinical diagnosis, genotyping, and other fields, thanks to its high accuracy and reliability, since its introduction in the 1970s. Although high-throughput sequencing technology has been widely used, Sanger sequencing is still the core technology in many laboratories and research projects because of its unique advantages in single-fragment high-precision sequencing (such as verification of cloned products, mutation detection, genotype confirmation, etc.).
In the Sanger sequencing process, experimental design and sample preparation are the key links to determine the success or failure of sequencing, which directly affects the quality and reliability of the final sequencing results. A scientific and reasonable experimental design can minimize interference factors and ensure efficient sequencing reaction. High-quality samples are the premise of obtaining clear and accurate sequencing maps. On the other hand, if the experimental design is flawed or the sample quality is not up to standard, it may lead to a weak sequencing signal, disordered peak pattern, fuzzy results, or even complete failure, which not only wastes time and resources but also may mislead the research conclusion.
This article details key points of Sanger sequencing experimental design, sample preparation methods for different types, and solutions to common problems, aiming to ensure the success and reliability of Sanger sequencing.
Key Points of Sanger Sequencing Experimental Design
The experimental design of Sanger sequencing is the core link to ensure the accuracy of the results. It needs to focus on the research goal, accurately control the three elements: primer design, template selection, and reaction conditions. Scientific design can reduce interference, improve reaction specificity and efficiency, and lay the foundation for high-quality sequencing results, which is the key prerequisite for the success of the experiment.
Selection and Design of Primers
Primers are the key components of the Sanger sequencing reaction, and their quality and applicability directly affect the specificity and efficiency of the sequencing reaction. The core principle of primer design is to ensure that it can specifically bind to template DNA, and at the same time avoid forming secondary structure (such as hairpin structure) or primer dimer, so as to ensure the smooth sequencing reaction.
In terms of primer length, it is usually recommended to design primers with 18-25 bases. Shorter primers (such as less than 15 bases) may lead to insufficient specificity, easy to combine with non-target sequences, and produce non-specific amplification or sequencing signal interference; However, too long primers (such as more than 30 bases) may increase the probability of secondary structure formation, reduce the binding efficiency with the template, increase the annealing temperature and increase the complexity of the reaction system.

Primer design process flow chart (Joshi et al., 2018)
Annealing temperature is another important parameter in primer design, and its level is mainly determined by GC content and the length of primers. Generally speaking, the annealing temperature can be roughly estimated by the formula (Tm=4×(G+C)+2×(A+T)). In actual reaction, the annealing temperature is usually set within the range of 2-5℃ above and below the Tm value.
In addition, the sequence of the 3′ end of the primer is very important for the extension reaction, so it is necessary to avoid the occurrence of continuous identical bases (especially G and C) to prevent the primer from mismatching or slipping. At the same time, the primer sequence should avoid being complementary to the repeated sequence and hairpin structure region in the template to reduce non-specific binding.
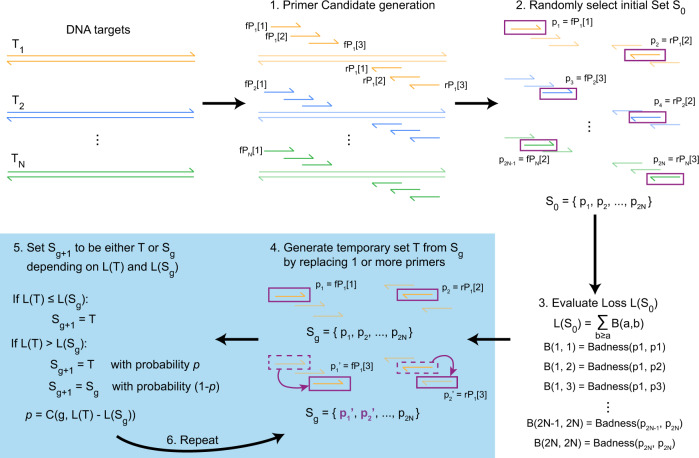
Overview of Simulated Annealing Design using Dimer Likelihood Estimation (Xie et al., 2022)
Determination of Template Type and Quality Requirements
The choice of template should be determined according to the research purpose and the characteristics of experimental materials. Common sequencing templates include plasmid DNA, PCR products, genomic DNA, cDNA, and so on.
- Plasmid DNA is one of the most commonly used sequencing templates, which is suitable for the verification of cloned fragments and the determination of gene insertion direction.
- Plasmid DNA as a template should have high purity and concentration, and the ratio of OD260/OD280 is usually required to be between 1.8 and 2.0 to avoid the interference of impurities such as protein and RNA.
- As a template, PCR products are suitable for direct sequencing of known fragments and detection of mutations. PCR products usually need to be purified to remove impurities before being used for sequencing, to ensure that the ratio of OD260/OD280 is about 1.8, and the concentration is generally recommended to be 10-50 ng/μL.
- Genomic DNA template requires complete fragments, no degradation, high purity (OD260/OD280=1.8-2.0), and the concentration is generally 50-100 ng/μL, depending on the sequencing purpose.
- cDNA template should pay attention to its reverse transcription efficiency and integrity to avoid the degradation of cDNA quality caused by RNA degradation.
Optimization of Sequencing Reaction Conditions
The optimization of sequencing reaction conditions is an important guarantee to obtain high-quality sequencing results, which mainly includes the determination of the reaction system, enzyme dosage, reaction temperature, and time.
The reaction system usually consists of template DNA, primers, dNTPs, dideoxynucleotide, DNA polymerase, buffer, and so on.
- The volume of the reaction system should be determined according to the experimental requirements and instrument specifications, and the common systems are 10 μL, 20 μL, and so on.
- In the system configuration, the ratio of template to primer is particularly critical, and it is generally recommended that the molar ratio of primer to template is 3:1-10:1.
- For plasmid template, because it is circular double-stranded DNA, the primer binding site is relatively single, and the primer dosage can be increased appropriately.
- For linear templates such as PCR products, the amount of primers can be adjusted according to the template concentration to avoid nonspecific signals caused by excessive primers.
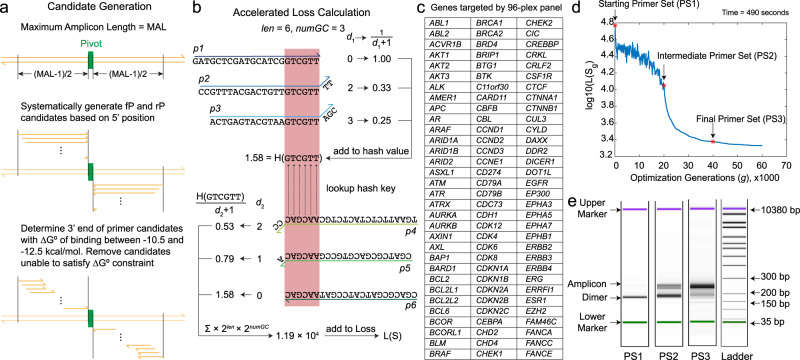
lmplementation and experimental evaluation of a multiplex primer designalgorithm based on the SADDLE framework (Xie et al., 2022)
The dosage of DNA polymerase should be determined according to the size of the reaction system and the complexity of the template. Excessive enzyme may lead to nonspecific extension and produce background signal. However, insufficient enzyme amount will reduce the extension efficiency and weaken the sequencing signal. Usually, 0.5-1U DNA polymerase is added to every 10 μL reaction system to meet the demand. For templates with high GC content or rich secondary structure, thermostable DNA polymerase (such as AmpliTaq DNA polymerase) with strong tolerance can be selected, and the amount of enzyme can be increased appropriately.
The setting of reaction temperature and time should follow three key steps of the sequencing reaction: denaturation, annealing, and extension.
- The denaturation step aims to untie the template DNA double strands, usually at 94-96℃ for 30 seconds to 1 minute.
- The temperature of the annealing step is determined according to the Tm value of the primer, which is generally 2-5℃ lower than the Tm value, and the time is 30 seconds to 1 minute, so as to ensure the specific binding between the primer and the template.
- The temperature of the extension step is usually 60-72℃, and the time depends on the expected sequencing length.
Generally, every 1kb fragment needs to be extended for about 1 minute. For long fragment sequencing, the extension time can be appropriately extended, but attention should be paid to avoid the decline of DNA polymerase activity.
In addition, the number of cycles is also an important factor affecting the sequencing reaction. Conventional sequencing reactions generally set 25-35 cycles. Too few cycles may lead to insufficient product quantity and a weak signal. Too many cycles may increase the accumulation of non-specific products and affect the peak quality.
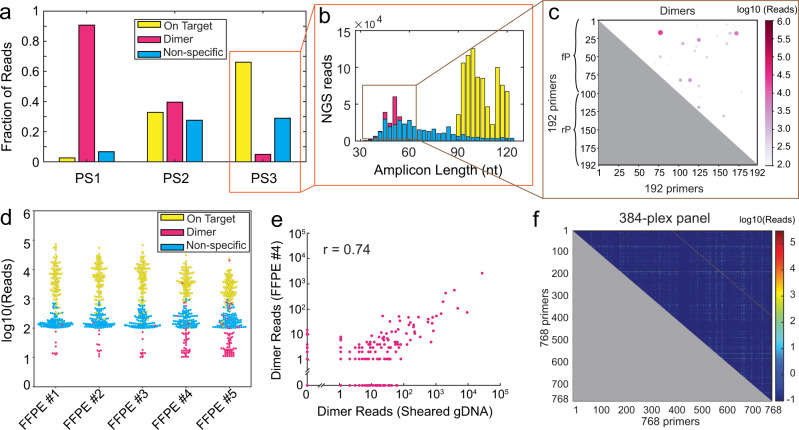
Experimental NGS results for SADDLE-designed primer sets (Xie et al., 2022)
Sanger Sequencing Sample Preparation
The accuracy of Sanger sequencing is highly dependent on sample quality, and sample preparation is the core link to determining the success or failure of sequencing. From genomic DNA, plasmid to PCR products, the extraction and purification of different types of samples need to follow strict standards, and their concentration, purity, and integrity directly affect the clarity of sequencing peaks and the reliability of results.
Genomic DNA
Genomic DNA extraction usually includes cell lysis, protein removal, DNA precipitation, and other steps.
- For animal tissue samples, protease K digestion combined with phenol-chloroform extraction method can be used: after cutting the tissue, add lysis buffer containing SDS, and digest it with protease K overnight at 55℃ to rupture the cells and release genomic DNA. Subsequently, it was extracted with phenol-chloroform mixed solution to remove impurities such as protein. The DNA in the upper water phase was precipitated with absolute ethanol, washed with 70% ethanol, and dissolved in TE buffer or ultrapure water.
- For blood samples, the difference between red blood cells and white blood cells can be used for processing. Red blood cell lysate was added to the whole blood sample, and white blood cells were collected by centrifugation after lysis of red blood cells, and then genomic DNA was extracted by the phenol-chloroform method mentioned above.
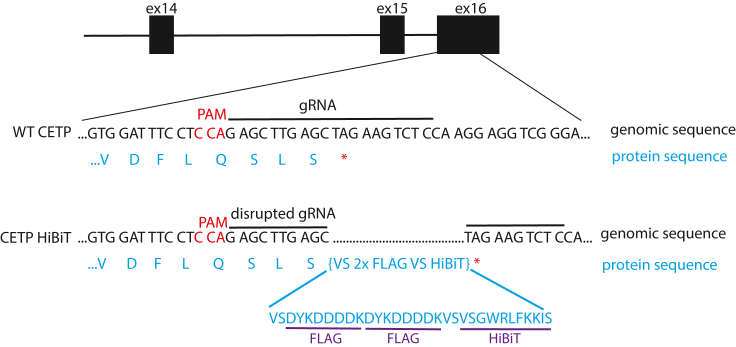
Schematic design of our CETP HiBiT editing strategy in human cells Presented is a Cterminal HiBiT tagging immediately prior to the CETP stop codon located in exon 16 (Lankford et al., 2024)
Plasmid DNA
The extraction methods of plasmid DNA mainly include alkali cracking and boiling, among which alkali cracking is the most commonly used. This method uses the difference in denaturation and renaturation between plasmid DNA and genomic DNA under alkaline conditions to separate plasmid DNA. The specific steps are as follows:
- Centrifuge bacterial culture containing plasmid to collect thalli, and add solution I (containing glucose and EDTA) to suspend thalli.
- Adding solution II (containing NaOH and SDS) to lyse the bacteria and denature the DNA.
- Adding solution III (potassium acetate buffer) to neutralize, renaturating plasmid DNA and forming a precipitate, while genome DNA and protein form a complex precipitate.
- After centrifugation, the supernatant was taken and further purified by phenol-chloroform extraction.
- Finally, plasmid DNA was obtained by ethanol precipitation.
cDNA
The extraction of cDNA needs to extract high-quality RNA from samples first, and then synthesize cDNA by reverse transcription. The commonly used methods of RNA extraction include the Trizol method and column extraction method.
- The Trizol method is suitable for a variety of tissue and cell samples. Cells are lysed by reagents such as guanidine isothiocyanate, which destroys the activity of RNase and protects RNA from degradation.
- Chloroform was added and centrifuged, RNA entered the water phase, precipitated by isopropanol, and washed with 70% ethanol to obtain total RNA.
- After obtaining high-quality total RNA, using it as a template, under the action of reverse transcriptase (such as M-MLV reverse transcriptase) and oligo (dT) or random primers as primers, the first strand of cDNA was synthesized.
The synthesized cDNA can be amplified by PCR to obtain the target fragment. After the amplified product is separated by agarose gel electrophoresis, it is purified by a gel recovery kit to remove the redundant primers, dNTPs, and other impurities, and the cDNA template, which can be used for sequencing, is obtained.
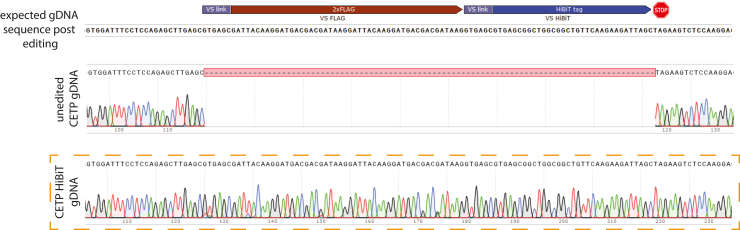
Example of Sanger sequencing results of HepG2 CETP HiBiT gDNA The expectedgDNA sequence of CETP exon 16 edited with insertion of the HiBiT sequence (VS 2xFLAG VSHiBiT) is shown for comparison at the top of this panel (Lankford et al., 2024)
Common Problems and Solutions in Sanger Sequencing
Although Sanger sequencing is mature and reliable, problems such as sample degradation, pollution, and abnormal signals often occur in the experiment, which directly affect the quality of the results. These problems mostly stem from improper sample preparation, unbalanced reaction conditions, or defects in primer design. Identifying the root cause of the problem in time and taking targeted solutions are the key to improving the success rate of sequencing and ensuring the reliability of data.
Degradation of Sample
Sample degradation is one of the common problems in sample preparation, which is characterized by DNA band dispersion, premature termination of the sequencing signal, or peak disorder. The main reasons leading to sample degradation are:
- The sample was not processed in time or stored improperly after collection, and the nuclease was not effectively inhibited.
- Improper operation in the extraction process, such as repeated freezing and thawing, violent oscillation, etc.
- The quality of the extraction reagent is poor or expired, which can not effectively protect DNA.
For the problem of sample degradation, we should first start with the link of sample collection and preservation, so as to ensure that the samples are processed or frozen immediately after collection to avoid the role of nuclease. In the extraction process, it is carried out in strict accordance with the operating procedures to avoid repeated freezing and thawing of samples, and the operation is gentle and reduces the mechanical damage to DNA.
Select high-quality extraction reagents and ensure that they are used within the validity period. If the sample has been slightly degraded, we can try to recover relatively complete DNA fragments by agarose gel electrophoresis for the sequencing reaction. For severely degraded samples, it is necessary to re-collect samples for extraction.
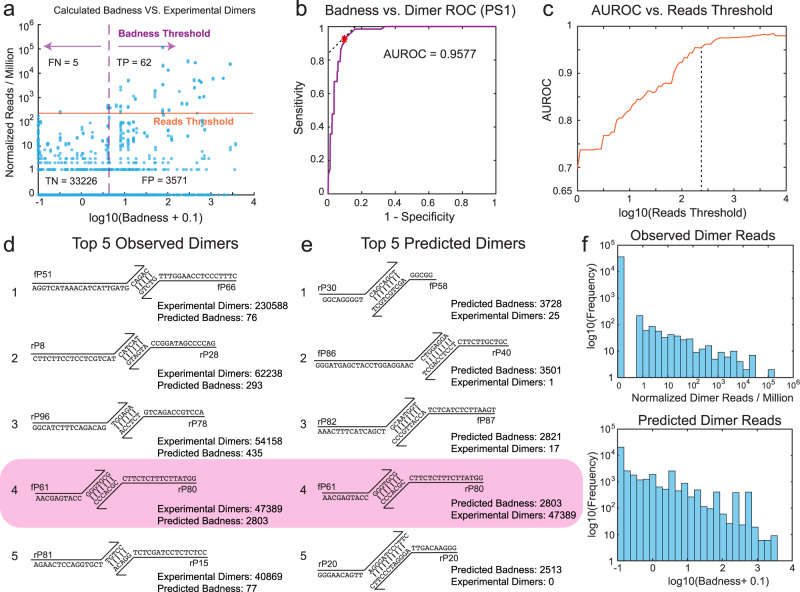
Evaluation of prediction accuracy of the Badness function for individual primerdimer candidates (Xie et al., 2022)
Sample Pollution
Sample pollution mainly includes exogenous DNA pollution, RNA pollution, and protein pollution, which will lead to peak, signal interference, or reaction inhibition in sequencing results.
- Exogenous DNA pollution may come from cross-contamination of experimental environment, operating instruments, or operators.
- RNA pollution is usually due to incomplete treatment of RNase during extraction.
- The pollution in protein may be due to imperfect extraction steps and incomplete removal of protein impurities.
In order to avoid foreign DNA pollution, the experimental operation should be carried out in a clean, ultra-clean workbench, and the operating instruments (such as centrifuge tubes and pipette tips) should be used with sterile disposable products. The experimenters should wear gloves and masks and disinfect the experimental environment regularly.
For RNA pollution, when extracting DNA, RNase A can be added to the extraction reagent to degrade RNA in the sample. If RNA pollution has occurred, an appropriate amount of RNase A can be added to the sample, incubated at 37℃ for 30 minutes, and then purified again.
For protein pollution, protein impurities can be removed by increasing the times of phenol-chloroform extraction or secondary purification with a column purification kit.
Insufficient Sample Concentration
Insufficient sample concentration will lead to insufficient template in the sequencing reaction and a weak signal, which will affect the accuracy of sequencing results. The reasons for the insufficient sample concentration may be:
- The low content of the sample itself (such as trace tissue samples).
- Excessive DNA loss during extraction, such as insufficient centrifugation in the precipitation step, DNA loss during washing, etc.
- The extraction reagent is inefficient and cannot effectively release DNA.
For the case of low sample content, the initial sample amount can be increased, or a more sensitive extraction method (such as the magnetic bead method) can be selected. In the process of extraction, optimize the precipitation steps, such as prolonging the centrifugation time and increasing the centrifugation speed to ensure the full precipitation of DNA. Be careful when washing to avoid bringing out DNA precipitation when absorbing supernatant. If the efficiency of the extraction reagent is low, a high-quality extraction reagent or kit can be replaced to improve the efficiency of DNA extraction.
For the sample with low concentration, a vacuum concentrator can be used to concentrate the sample to improve its concentration to the level required for sequencing, but attention should be paid to avoid excessive concentration, leading to an increase in impurity concentration in the sample and affecting the sequencing reaction.
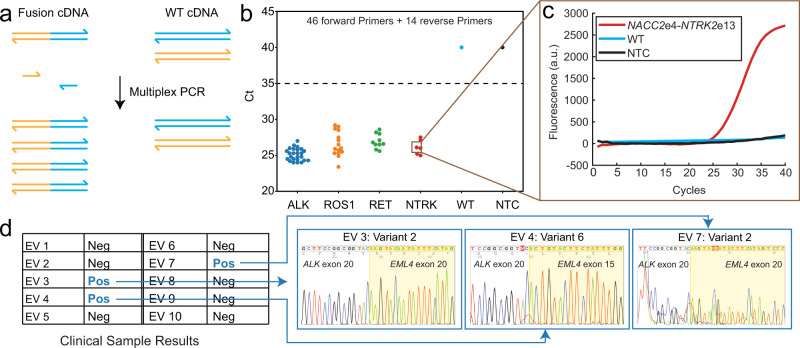
Highly multiplexed qPCR detection of gene fusions using SADDLE-designed primer sets (Xie et al., 2022)
Conclusion
The accuracy of Sanger sequencing depends on rigorous experimental design and standardized sample preparation. From the detailed control of primer design, the scientific selection of template type, to the strict operation of sample extraction and purification, every step is related to the reliability of the final result. Following the above points and avoiding common problems can significantly improve the efficiency and quality of sequencing, provide solid technical support for scientific research and clinical diagnosis, and promote the in-depth application of gene sequencing technology in various fields.
Learn More
References
- Joshi AP, Angel A, Angel B, et al. "In-silico Designing and Testing of Primers for Sanger Genome Sequencing of Dengue Virus Types of Asian Origin." J Genomics. 2018 6: 34-40 https://doi.org/10.7150/jgen.22460
- Sikkema-Raddatz B, Johansson LF, de Boer EN, et al. "Targeted next-generation sequencing can replace Sanger sequencing in clinical diagnostics." Hum Mutat. 2013 34(7): 1035-1042 https://doi.org/10.1002/humu.22332
- Xie NG, Wang MX, Song P, et al. "Designing highly multiplex PCR primer sets with Simulated Annealing Design using Dimer Likelihood Estimation (SADDLE)." Nat Commun. 2022 13(1): 1881 https://doi.org/10.1038/s41467-022-29500-4
- Lankford KP, Hulleman JD. "Protocol for HiBiT tagging endogenous proteins using CRISPR-Cas9 gene editing." STAR Protoc. 2024 5(2): 103000 https://doi.org/10.1016/j.xpro.2024.103000