
 Sample Submission Guidelines
Sample Submission Guidelines
Population Evolution
What is the Population Evolution Theory
Population evolution theory scrutinizes the dynamic shaping of genetic variation within populations over time, driven by a suite of evolutionary processes. This sophisticated theory amalgamates principles from genetics, ecology, and evolutionary biology to elucidate how populations adapt to their environments, undergo evolutionary transformations, and potentially diverge into new species.
The theory hinges on several fundamental mechanisms, which are pivotal to understanding population evolution:
- Natural Selection: This cornerstone process encompasses the differential survival and reproduction of individuals based on phenotypic variations. Over successive generations, traits that confer survival and reproductive advantages become increasingly common within the population, thereby directing its evolutionary pathway.
- Genetic Drift: Particularly pronounced in small populations, genetic drift refers to random fluctuations in allele frequencies. This stochastic process can result in significant evolutionary changes, including the fixation or loss of alleles, regardless of their selective value. Consequently, genetic drift can lead to divergent evolutionary outcomes independent of natural selection.
- Gene Flow: Also known as migration, gene flow entails the transfer of alleles between populations. This movement of genetic material can introduce novel alleles into a population, enhancing genetic diversity and impacting its evolutionary course. Gene flow serves as a conduit for genetic exchange, which can either constrain or promote divergence among populations.
- Mutation: Mutations, defined as alterations in DNA sequences, serve as the primary source of new genetic variations. These genetic changes can foster the emergence of new traits, providing the fundamental raw material needed for natural selection and other evolutionary processes to act upon. Consequently, mutations are indispensable for the continuous evolution and adaptive potential of populations.
Introduction to Population Evolution Analysis
Population evolution analysis entails the application of cutting-edge genomic technologies to study the genetic composition and evolutionary pathways of populations. Leveraging data obtained from high-throughput sequencing techniques, this analysis reveals genetic variations such as single nucleotide polymorphisms (SNPs), insertions and deletions (InDels), structural variations (SVs), and copy number variations (CNVs).
Key Components of Population Evolution Analysis
- Sample Collection and Preparation: The foundation of a robust analysis lies in the meticulous selection of representative samples from various subpopulations or ecological niches. Ensuring the quality and diversity of these samples is essential for yielding accurate and meaningful results.
- Genomic Sequencing: Utilizing advanced sequencing technologies, such as whole-genome sequencing (WGS) and targeted resequencing, researchers can gather extensive genomic data. For instance, CD Genomics employs both second-generation (Illumina) and third-generation (PacBio and Nanopore) sequencing platforms to extract detailed genetic information, facilitating comprehensive analysis.
- Variant Detection: Identifying genetic variations within populations is a cornerstone of understanding evolutionary processes. Methods for variant detection include SNP calling, InDel detection, and the analysis of SVs and CNVs. These approaches are pivotal for mapping genetic diversity and tracing evolutionary histories.
- Bioinformatics Analysis: Computational tools and statistical models play a crucial role in interpreting genetic data. This involves determining population structure and identifying selection signatures through bioinformatics software designed for variant annotation, population genetics metrics, and evolutionary modeling. Such tools enable a deeper understanding of the genetic underpinnings and evolutionary dynamics within populations.
Advantages of Population Evolution Analysis
- Comprehensive Genetic Insights: Reveals the genetic basis of adaptation and speciation by analyzing genetic variation across diverse populations.
- Enhanced Understanding of Evolutionary Dynamics: Tracks genetic changes over time to uncover migration patterns, adaptation mechanisms, and the impact of historical events.
- Practical Applications in Conservation and Breeding: Aids in identifying genetically diverse individuals for breeding and conservation, ensuring genetic health and resilience.
- Advanced Technological Integration: Utilizes cutting-edge technologies and bioinformatics tools, with CD Genomics providing precise and high-quality data for in-depth research.
- Extensive multiplexing flexibility and high-throughput sequencing, enables for detecting large number of SNPs, InDels, CNVs, CVs.
- Time and cost efficient.
- Dedicate support from specialized PhD-level scientists.
Applications of Population Evolution
- Research on the mechanism of artificial domestication: The genetic relationship of wild-type and domesticated populations was inferred through genetic analysis of the two populations, then key genes related to important economics traits were identified, which provided good guidance for the agricultural breeding of animals and plants.
- Analysis of natural selection mechanism: The genes selected in the adaptive evolution process can be unearthed through the research of populations from different geographical areas, then providing environmental adaptive genetic resources for breeding work.
- Population history research: By analyzing the possible origin of the species and genetic variation information of the population in each distribution area, the evolution process of the species can be explored.
Population Evolution Workflow
CD Genomics provides population evolution services that include sample collection, DNA sequencing, and comprehensive bioinformatics analysis. These services assess genetic variation, adaptation mechanisms, and evolutionary dynamics. Through detailed data interpretation, they offer critical insights into population history, genetic diversity, and conservation strategies, supporting efforts to maintain genetic health and biodiversity.

Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
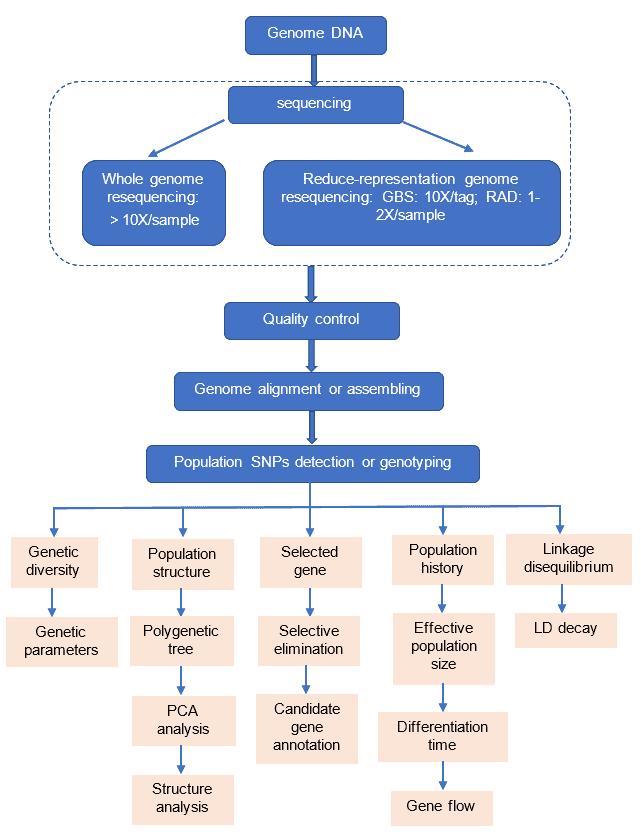
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Population Evolution for your writing (customization)
References
- Nadeau N J, Ruiz M, Salazar P, et al. Population genomics of parallel hybrid zones in the mimetic butterflies, H. melpomene and H. erato. Genome Research. 2014, 24(8): 1316-1333.
- Tine M, Kuhl H, Gagnaire P A, et al. European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nature communications, 2014, 5.
Demo Results
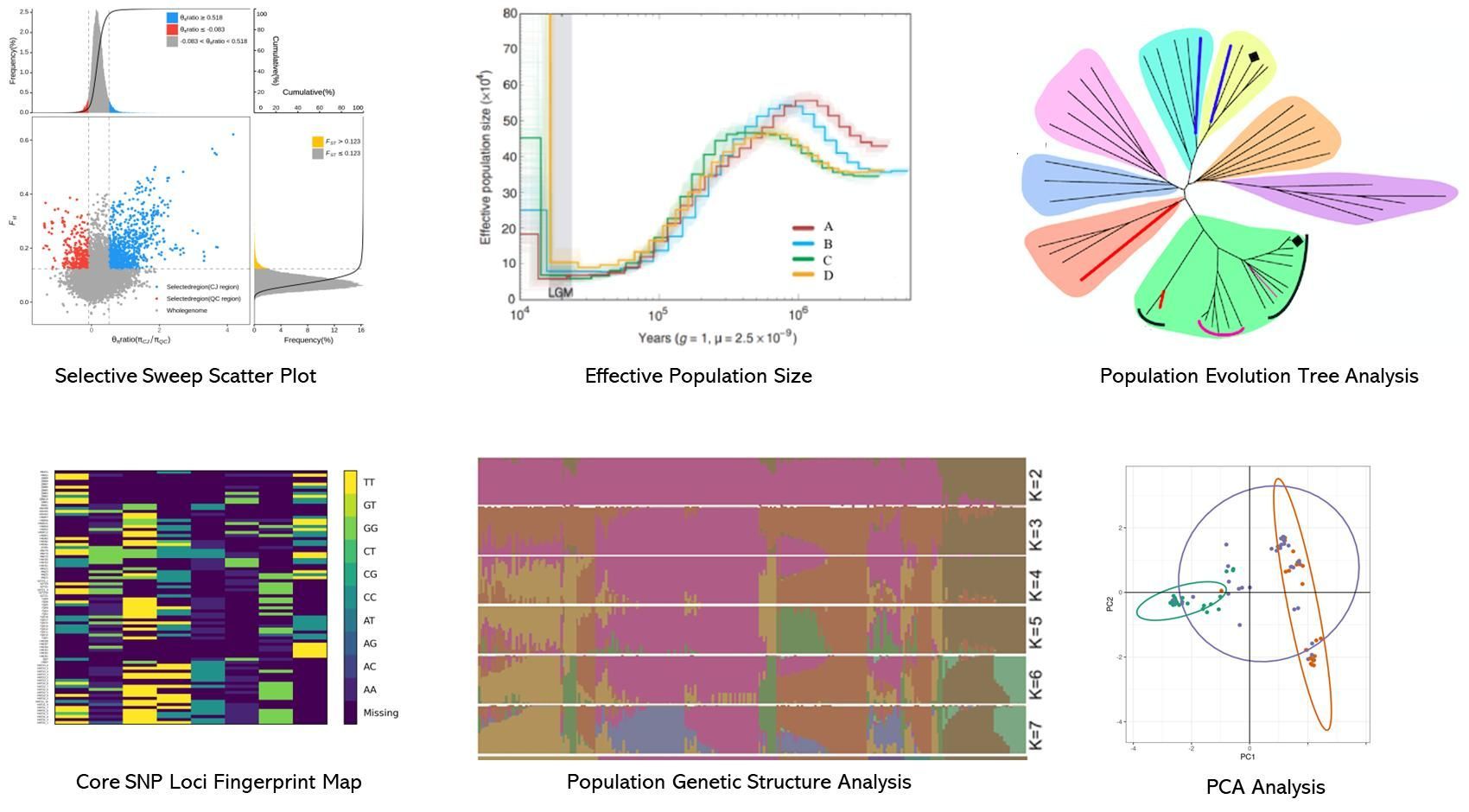
Partial results are shown below:
Population Evolution FAQs
1. How can subpopulation differentiation be inferred?
Subpopulation differentiation can be inferred through meticulous analysis of geographical and environmental variations. Prolonged geographical isolation or significant environmental disparities frequently result in the formation of distinct subpopulations. Additionally, factors such as genetic drift and selective pressures play crucial roles in promoting differentiation, leading to unique genetic signatures within each subpopulation.
2. What is the recommended sequencing depth for population evolution studies?
To obtain precise and comprehensive results in population evolution studies, a sequencing depth of at least 10X is recommended. However, higher depths, such as 30X or 50X, may be warranted for specific analyses, such as the detection of structural variants or detailed evolutionary investigations. These increased depths enhance the accuracy of genetic variation identification and improve the reliability of the study's findings.
3. What criteria should be used for selecting research populations?
The selection of research populations should be based on clear and distinct subpopulation characteristics, such as the differences between wild and domesticated types or populations originating from diverse geographical regions. It is imperative to acquire representative samples from each subpopulation to ensure the accuracy and validity of the analysis. Establishing clear subpopulation distinctions enhances our understanding of genetic variation and evolutionary dynamics.
Population Evolution Case Studies
Analysis of 427 genomes reveals moso bamboo population structure and genetic basis of property traits
Journal: Nature Communications
Impact factor: 17.7
Published: 15 September 2021
Background
Moso bamboo (Phyllostachys edulis), covering 74% of global bamboo areas and vital to China's economy, faces growth challenges due to environmental and human impacts. Whole-genome resequencing (WGRS) of 427 samples from 15 regions helps uncover genetic variations, providing insights into its evolutionary history and traits relevant to bamboo properties.
Materials & Methods
Sample Preparation
- Moso bamboo
- Young leaves
- DNA extraction
Sequencing
- Whole-genome resequencing
- Illumina sequencing platform
- SNP and InDel calling
- SV and CNV detection
- The related gene analysis
- Phylogeny construction
- Population structure analysis
- Genome-wide association study
Results
Large-scale whole-genome resequencing (WGRS) of 427 moso bamboo samples from 15 regions revealed low genomic diversity, with an average SNP density of one per 351 base pairs. Most SNPs were found in intergenic regions, with a higher nonsynonymous-to-synonymous substitution ratio compared to other plants. This low diversity suggests a small effective population size and limited genetic pool for future breeding.
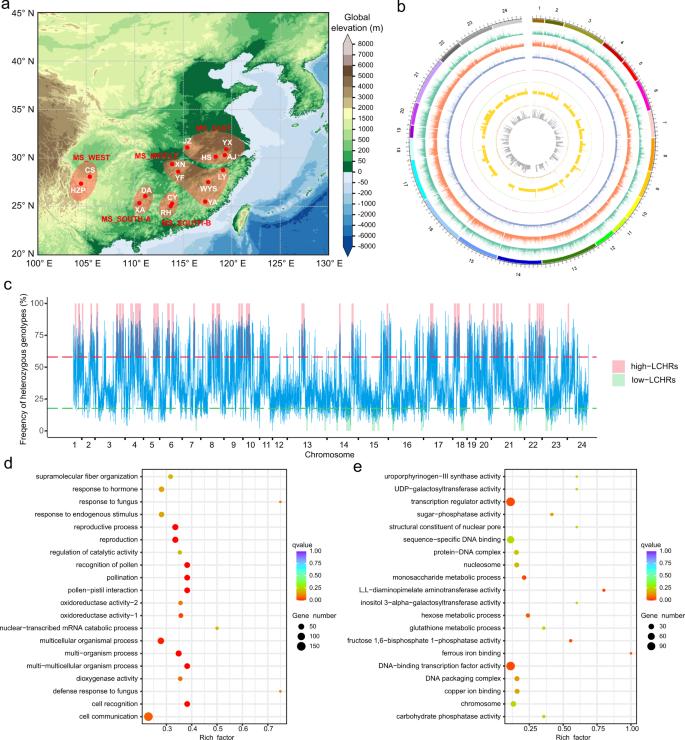 Fig. 1: The landscape of sampling and variants in sequenced moso bamboo individuals.
Fig. 1: The landscape of sampling and variants in sequenced moso bamboo individuals.
Balancing selection plays a key role in moso bamboo's adaptation to its environment, maintaining genetic diversity despite low overall divergence. Analysis identified 83 significant genomic regions related to balancing selection, with genes involved in disease resistance and environmental responses showing high-frequency variations. This suggests that balancing selection helps sustain genetic diversity and supports adaptation.
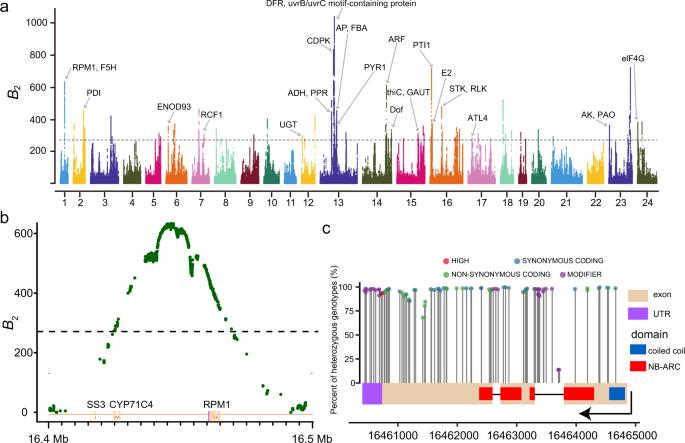 Fig. 2: Balancing selection in the moso bamboo population underlies adaptation.
Fig. 2: Balancing selection in the moso bamboo population underlies adaptation.
A GWAS of moso bamboo revealed genetic variants associated with traits like culm height and mechanical strength. Analyzed SNPs identified significant markers linked to cell wall properties and environmental adaptation. Key genes, such as cinnamoyl-CoA reductase, impact traits like lignin levels, providing insights for breeding and genetic research.
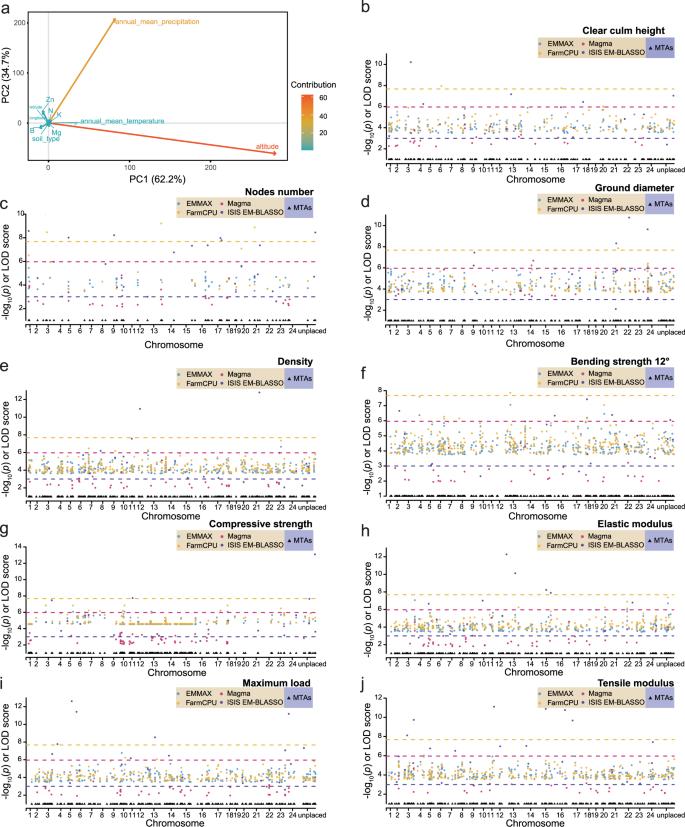 Fig. 3: GWAS of important property traits.
Fig. 3: GWAS of important property traits.
Conclusion
Moso bamboo's whole-genome sequencing revealed low genetic diversity but high heterozygosity due to asexual reproduction. Conservation should focus on diverse populations, and understanding genetic variations can enhance breeding and sustainable forest management.
Reference
- Zhao H, Sun S, Ding Y, et al. Analysis of 427 genomes reveals moso bamboo population structure and genetic basis of property traits. Nature Communications. 2021, 12(1):5466.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Collection of genetic data in ethnic-based studies across Aymaras, Quechuas and Mestizos: the challenges of the Genetics of Alzheimer's in Peruvian Population (GAPP) study
Journal: Alzheimer's & Dementia
Year: 2022
Evaluation of Plasma Biomarkers for A/T/N Classification of Alzheimer Disease Among Adults of Caribbean Hispanic Ethnicity
Journal: JAMA Network Open
Year: 2023
Increased Production of Pathogenic, Airborne Fungal Spores upon Exposure of a Soil Mycobiota to Chlorinated Aromatic Hydrocarbon Pollutants
Journal: Microbiology Spectrum
Year: 2023
A Splice Variant in SLC16A8 Gene Leads to Lactate Transport Deficit in Human iPS Cell-Derived Retinal Pigment Epithelial Cells
Journal: Cells
Year: 2021
See more articles published by our clients.


