
 Sample Submission Guidelines
Sample Submission Guidelines
2'-O-RNA Methylation Sequencing Service
CD Genomics offers state-of-the-art 2'-O-RNA Methylation Sequencing services, specifically designed to detect and analyze 2'-O-methylation patterns in RNA molecules. Our methodology provides high sensitivity and accuracy, enabling precise identification of methylated nucleotides. This service supports in-depth studies on RNA structure, stability, and regulatory roles in biological processes.
What is 2'-O methylation of RNA
2'-O-Methylation (Nm) is a post-transcriptional modification of RNA, catalyzed by 2'-O-methyltransferases, which involves the substitution of a hydrogen atom on the 2-hydroxyl group with a methyl group. This chemical modification occurs at the 2' position of ribose in RNA and is mediated by RNA methyltransferases or fibrillarin. Notably, 2'-O-RNA methylation is a modification found ubiquitously across various species, including mammals, yeast, plants, and viruses.
Recent studies have demonstrated that 2'-O-RNA methylation is present on diverse RNA molecules such as mRNA, tRNA, rRNA, and miRNA. Structurally, this modification enhances the hydrophobicity of the nucleotide, thereby increasing its stability. Functional implications of 2'-O-RNA methylation have been elucidated in multiple biological processes; it affects mRNA-protein interactions, regulates rRNA translation efficiency, and participates in tRNA recognition.
Transfer RNA (tRNA) harbors an extensive array of post-transcriptional modifications crucial for exerting its biological functions. Among these, methylation stands out as the most prevalent type of tRNA modification, occurring either on the nucleotide bases or at the 2'-O-ribose position (2'-O-methylation). The 2'-O-methylation modification is widespread in archaea, prokaryotes, and eukaryotes, identified at positions 4, 6, 18, 32, 34, 39, 44, 54, and 56 within tRNA.
Research indicates that 2'-O-methylation plays a regulatory role in tRNA folding, maturation, stability, and the precision of anticodon-mRNA codon pairing. It is closely associated with cellular processes such as growth, stress response, and immune regulation. Deficiencies in cytoplasmic tRNA 2'-O-methylation are often linked to various diseases. Enzymes involved in tRNA 2'-O-methylation present potential targets for drug development.
Investigating tRNA 2'-O-methylation and its modifying enzymes will enhance our understanding of the biological functions of this modification. Furthermore, it lays the groundwork for exploring the pathogenic mechanisms of tRNA 2'-O-methylation enzymes in human diseases.
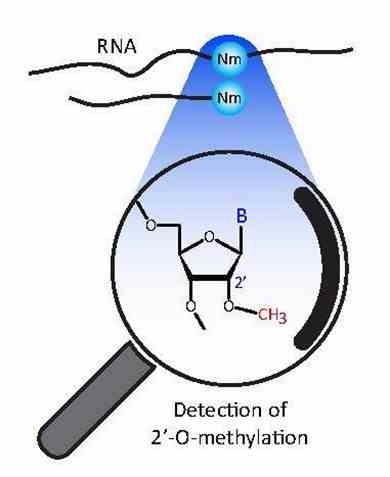 Figure 1. Detection of RNA Ribose 2'-O-Methylations (Yuri Motorin, Virginie Marchand, 2018)
Figure 1. Detection of RNA Ribose 2'-O-Methylations (Yuri Motorin, Virginie Marchand, 2018)
The Introduction of 2'-O-RNA Methylation Sequencing
To detect more 2'-methylation modifications and elucidate further biological functional mechanisms, numerous biological experimental techniques have been proposed. These include RNase H-based methods, reverse transcription-based approaches, and PCR-based strategies. However, these methods are often time-consuming. With the advancement of sequencing technologies, the accumulation of nucleotide sequences continues. The core of 2'-O-RNA methylation sequencing lies in identifying the 2'-O-methylation sites within RNA molecules using chemical or enzymatic methods, followed by precise localization and quantification of these sites through high-throughput sequencing techniques. The 2'-O-methylation alters the chemical properties of RNA molecules, enabling the identification and localization of these modifications.
Currently, several methodologies are predominantly employed for 2'-O-RNA methylation sequencing:
- Ribose-seq: This technique focuses on ribosomal RNA (rRNA). It involves the targeted enzymatic cleavage of RNA chains to release 2'-O-methylated ribose, which is subsequently subject to high-throughput sequencing.
- Nm-seq: This method employs specific chemical modifications to label 2'-O-methylation sites. These modifications allow for the recognition of methylation sites through reverse transcription reactions followed by high-throughput sequencing.
- Pseudouridine-seq (Ψ-seq): Although primarily designed to identify pseudouridine (Ψ) modifications, this method can be integrated with other detection techniques to facilitate the study of 2'-O-methylation.
CD Genomics offers a comprehensive 2'-O-RNA methylation detection service, designed to identify the presence of 2'-O-methylation modifications on RNA molecules and determine the specific sites of 2'-O-methylation. Beyond the offered service, CD Genomics has innovated a suite of products designed for the assessment of 2'-O-RNA methylation levels in diverse RNA molecules, encompassing mRNA, LncRNA, pri-miRNA, tRNA, and rRNA.
Deep Sequencing-Based Approaches for Detection of 2'-O-methylation
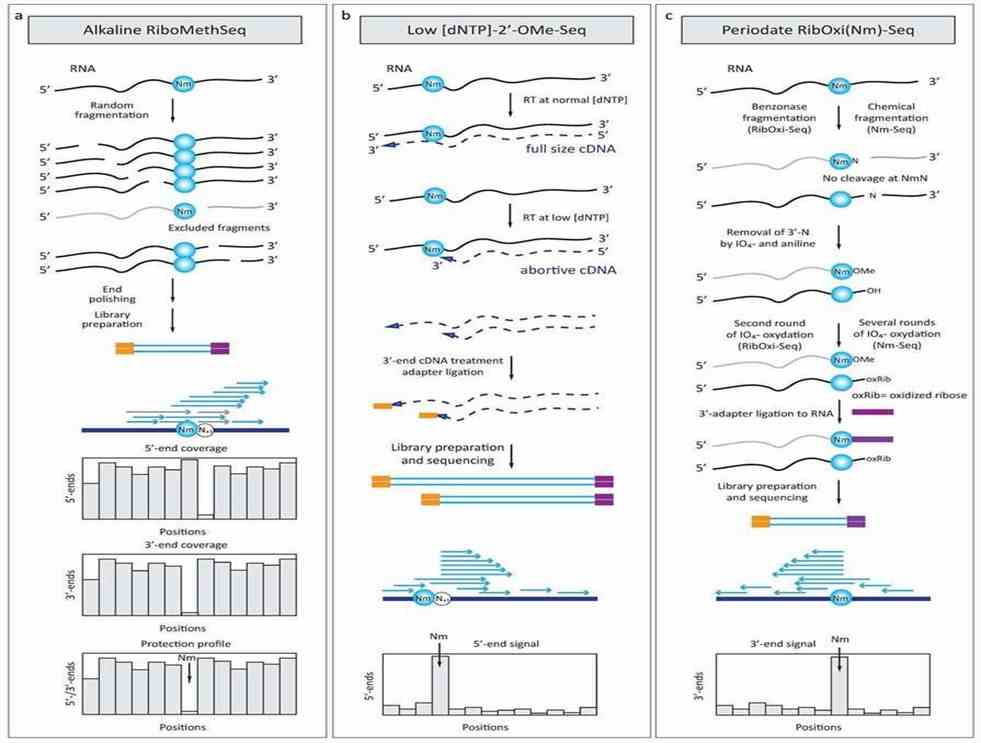 Figure 2. Deep sequencing-based approaches for detection of 2'-O-methylated residues in RNA (Yuri Motorin, Virginie Marchand, 2018)
Figure 2. Deep sequencing-based approaches for detection of 2'-O-methylated residues in RNA (Yuri Motorin, Virginie Marchand, 2018)
Advantages of Our 2'-O-RNA Methylation Sequencing Service
- Individual Nucleotide Precision: Our service excels in pinpointing 2'-O-RNA methylation modifications with single-nucleotide precision. This sophisticated approach guarantees a meticulous examination of modification locations within RNA molecules.
- Efficient High-Throughput Detection: Employing advanced high-throughput techniques, we ensure the parallel and efficient identification of 2'-O-RNA modification sites throughout the entire transcriptome. This strategy allows for the simultaneous evaluation of multiple RNA molecules, furnishing a comprehensive overview of 2'-O-RNA methylation patterns.
- Comprehensive Coverage Across RNA Species: Our service spans a diverse array of RNA types, encompassing mRNA, LncRNA, pri-miRNA, tRNA, and rRNA. This extensive analysis guarantees the identification of 2'-O-RNA methylation sites across various RNA molecules, contributing to a nuanced comprehension of the modification landscape.
- Specialized Bioinformatic Interpretation: Supported by an adept bioinformatics team, we deliver specialized data analysis services tailored to diverse customer requirements. Our team is proficient in conducting thorough data analyses, offering insightful interpretations of results derived from 2'-O-RNA methylation detection experiments.
Applications of 2'-O-RNA Methylation Sequencing
2'-O-RNA methylation sequencing has important applications in various research fields, including:
- RNA Biology Research: Studying the roles of 2'-O-methylation in RNA function, stability, and translation regulation.
- Disease Research: 2'-O-methylation is associated with various diseases (e.g., cancer, neurodegenerative diseases). Sequencing technology can reveal the mechanisms of these modifications in diseases.
- Drug Development: Identifying targets related to 2'-O-methylation provides a basis for new drug development.
2'-O-RNA Methylation Sequencing Workflow
CD Genomics leverages advanced sequencing platforms and years of experience to offer comprehensive 2'-O-RNA methylation detection services. These services primarily include sample processing, sequencing, and subsequent bioinformatics analysis.

Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis
We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
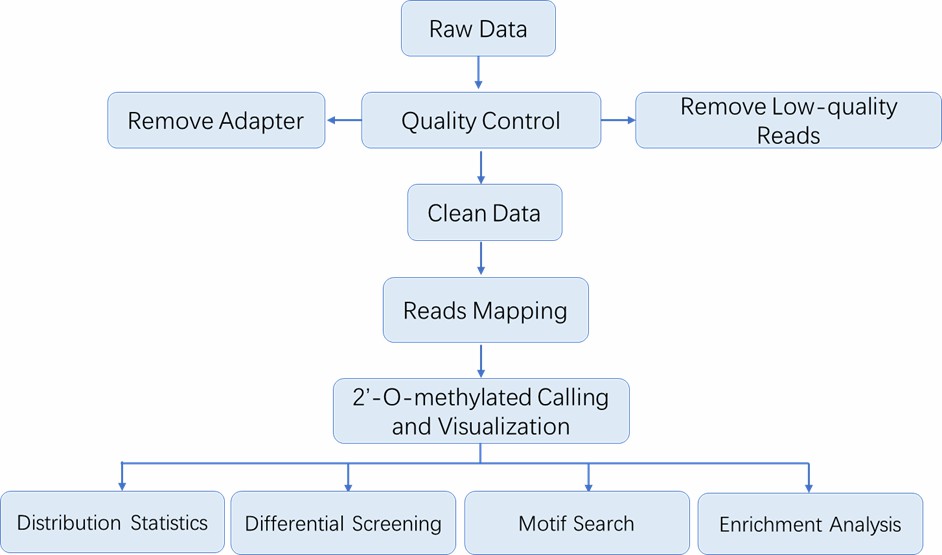
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in 2'-O-RNA Methylation Sequencing for your writing (customization)
Explore RNA modifications with CD Genomics' 2'-O-RNA Methylation Sequencing services. We provide advanced sample processing, precise sequencing, and comprehensive bioinformatics analysis to identify and quantify 2'-O-methylation sites. Contact us to enhance your research with detailed insights into RNA methylation.
Reference
- Motorin Y, Marchand V. Detection and analysis of RNA ribose 2'-O-methylations: challenges and solutions. Genes, 2018, 9(12): 642.
Demo Results
Partial results are shown below:
Reference
- Dai Q, Moshitch-Moshkovitz S, Han D, et al. Nm-seq maps 2'-O-methylation sites in human mRNA with base precision. Nature methods, 2017, 14(7): 695-698.
2'-O-RNA Methylation Seq FAQs
1. Why is rRNA Methylated on Selected 2' OH?
rRNA (ribosomal RNA) undergoes methylation on selected 2'-OH groups due to several critical reasons:
Structural Stability: 2'-O-methylation contributes significantly to the structural integrity of rRNA. This modification plays a pivotal role in preserving the correct three-dimensional conformation of rRNA, which is fundamental for the proper assembly and functionality of the ribosome.
Resistance to Degradation: Methylation at the 2'-OH position enhances the resistance of rRNA to ribonucleases, enzymes responsible for RNA degradation. This increased resistance is essential for maintaining the longevity and function of rRNA within the ribosome.
Accuracy of Translation: 2'-O-methylation is crucial for the fidelity of protein synthesis. The modified nucleotides aid in the accurate positioning of tRNAs and mRNA during translation, thereby minimizing errors in protein synthesis.
Interaction with Ribosomal Proteins: Methylated rRNA exhibits enhanced interactions with ribosomal proteins and other translation-related factors. These interactions are vital for the assembly and stability of the ribosomal complex.
Functional Sites: Methylation occurs at specific sites that are crucial for ribosomal activity. These modifications can affect the catalytic functions of the ribosome and its interactions with other molecules, thereby ensuring efficient and effective protein synthesis.
2. What types of RNA can be analyzed for 2'-O-methylation?
This sequencing method can be applied to various types of RNA, including:
- Messenger RNA (mRNA)
- Ribosomal RNA (rRNA)
- Transfer RNA (tRNA)
- Small nuclear RNA (snRNA)
- Long non-coding RNA (lncRNA)
3. Why is 2'-O-RNA methylation important?
2'-O-RNA methylation holds significant importance for various cellular processes, encompassing:
- Enhancement of RNA Stability and Degradation Resistance: This modification confers increased stability to RNA molecules, protecting them against degradation by ribonucleases and other enzymatic activities that could otherwise compromise RNA integrity.
- Maintenance of Proper RNA Secondary and Tertiary Structures: By influencing the folding and structural conformations of RNA, 2'-O-methylation ensures that RNA molecules adopt configurations essential for their biological functions.
- Regulation of RNA-Protein Interactions: This methylation facilitates specific and high-affinity interactions between RNA molecules and RNA-binding proteins, which are vital for various RNA-mediated regulatory mechanisms.
- Accuracy and Efficiency in Translation and Splicing: 2'-O-methylation aids in the precise and effective execution of translation and splicing processes, minimizing errors and enhancing the overall fidelity and efficiency of protein synthesis and RNA processing.
4. How do I prepare my samples for 2'-O-RNA methylation sequencing?
To prepare samples:
- Use fresh or properly stored tissues or cells.
- Follow recommended RNA extraction protocols to obtain high-quality RNA.
- Avoid RNase contamination to prevent RNA degradation.
5. How long does the 2'-O-RNA methylation sequencing process take?
The turnaround time depends on various factors, including the number of samples, the complexity of the analysis, and the service provider. Typically, the process can take several weeks from sample submission to data delivery.
2'-O-RNA Methylation Seq Case Studies
Case Study 1:
Nm-seq maps 2'-O-methylation sites in human mRNA with base precision
Journal: Nature Methods
Impact factor: 26.9
Published: 28 February 2018
Background
mRNA modifications, part of the epitranscriptome, regulate gene expression and include 2'-O-methylation (Nm). Nm is abundant in rRNA, tRNA, snRNA, and microRNA, essential for their function and stability. Traditional mapping methods, such as 2OMe-seq and RiboMeth-seq, identify Nm sites by detecting pauses in reverse transcription or resistance to hydrolysis. Existing methods struggle with rare RNA or low stoichiometry Nm sites. The new Nm-seq method uses oxidative cleavage to map Nm sites with high sensitivity and single-nucleotide precision.
Materials & Methods
Sample Preparation
- Human HeLa (cervical adenocarcinoma)
- HEK293 (human embryonic kidney) cells
- Total RNA extraction
- RNA purification
Sequencing
- Nm-seq
- Illumina HiSeq2500
- LC-MS/MS
- Identification of Nm sites in rRNA
- Identification of Nm sites in the transcriptome
- Motif search
- Metagene profiling
- GO enrichment analysis
- Statistical analysis
Results
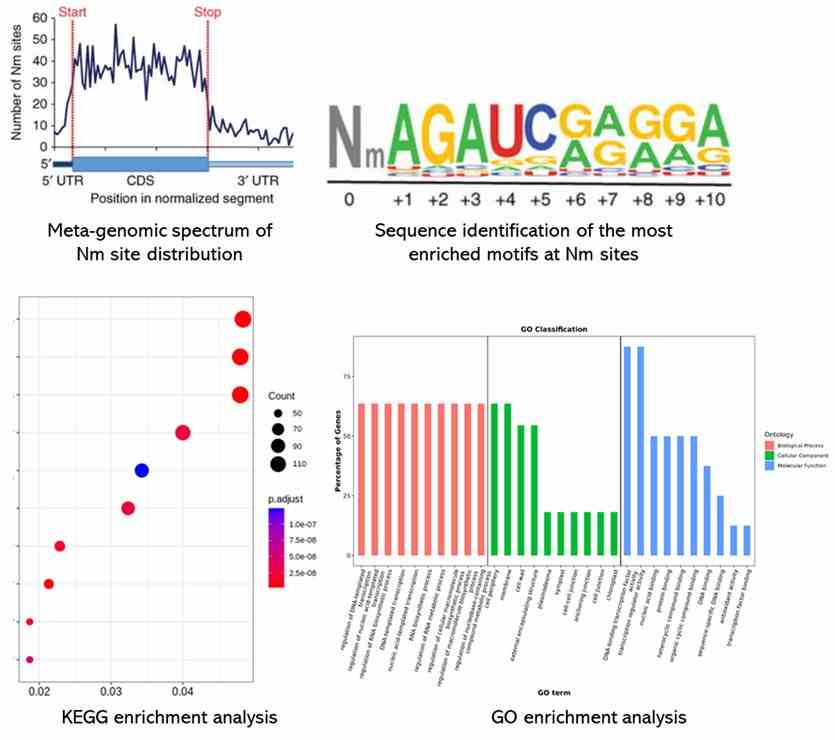
This article presents, for the first time, the identification of thousands of methylation sites on human Hela and 293T cell mRNA and describes the distribution of these methylation sites. Among them, the majority of methylation sites (95.7%) are located within 2398 RefSeq annotated genes, with 95.9% occurring in protein-coding genes, and a small fraction in non-coding genes (4.1%). Within protein-coding transcripts, the data reveals that the majority of sites are located in the coding sequence (CDS) region (70.3%). Motif analysis indicates that the signature sequence for 2'-O-methylation is AGAU, followed by a high distribution of A or G bases. Furthermore, 2'-O-methylation sites are enriched in protein-coding codons for three amino acids.
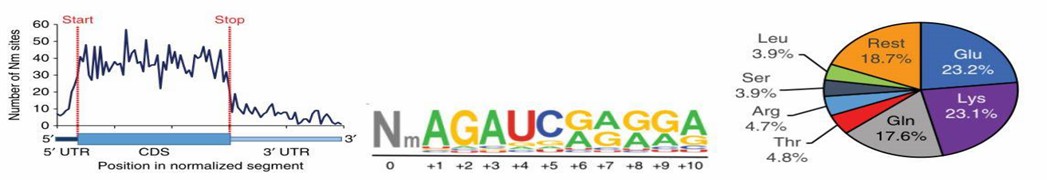 Figure 1: Characteristics of 2'-O-RNA Methylation Sites on Human Cell mRNA Molecules.
Figure 1: Characteristics of 2'-O-RNA Methylation Sites on Human Cell mRNA Molecules.
Conclusion
In summary, this study unveils, for the first time, the distribution characteristics of 2'-O-methylation in human cells.
Case Study 2: Involvement of 2'-O-RNA Methyltransferase FTSJ3 in Innate Immune Regulation of HIV
Recently, the research group led by Professor Yamina Bennasser at the University of Montpellier in France discovered, for the first time, the presence of 2'-O-methylation sites on the HIV virus. These sites exhibit activity in host cell infection and are regulated by the methyltransferase FTSJ3.
FTSJ3 as a 2'-O-Methyltransferase:
Through mass spectrometry and Western blot experiments, it was observed that the methylation reader protein TRBP interacts with the methyltransferase FTSJ3, forming the TRBP-FTSJ3 complex.
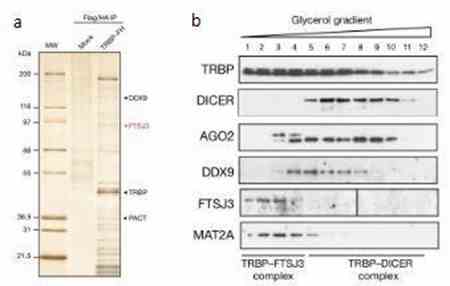 Figure 2: Formation of Two Distinct Complexes by TRBP.
Figure 2: Formation of Two Distinct Complexes by TRBP.
2'-O-Methylation Modification in HIV-1 RNA Viral Particles:
Researchers employed 2'-O-methylation sequencing to examine the methylation status of HIV-1 RNA. They identified 17 2'-O-methylation sites, with 15 of them located on adenine. Subsequent methylation sequencing of HIV-1 packaged in FTSJ3-knockout cells revealed a reduction in methylation levels at multiple sites. This indicates the influence of the methyltransferase FTSJ3 on the 2'-O-methylation pattern.
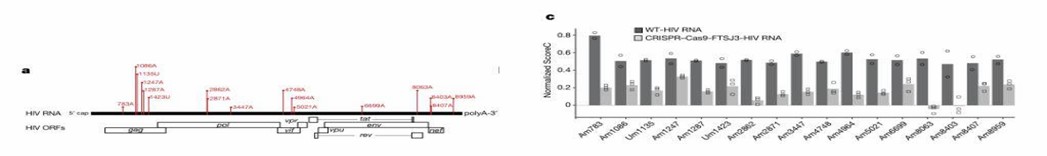 Figure 3: Methylation Profile of HIV-1 RNA and the Impact of FTSJ3 Depletion.
Figure 3: Methylation Profile of HIV-1 RNA and the Impact of FTSJ3 Depletion.
FTSJ3 Involvement in the Immune Regulation Mechanism of HIV-1:
Effect on IFN-α/IFN-β Expression: Firstly, does intracellular FTSJ3 influence the infection of host cells by the virus? The researchers used U937 monocytic cells expressing HIV RNA, with either wild-type (WT) or FTSJ3-siRNA knockdown. The results revealed a significant increase in the expression of IFN-α/IFN-β in the experimental group compared to the WT group.
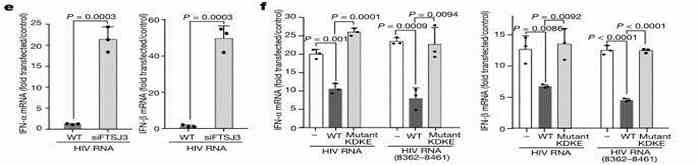 Figure 4: Involvement of FTSJ3 in the Immune Regulation Process of HIV-1.
Figure 4: Involvement of FTSJ3 in the Immune Regulation Process of HIV-1.
MDA5 Regulation of Methyltransferase in the Immune Modulation of HIV-1:
MDA5, acting as a cytoplasmic RNA sensor, plays a crucial role in immune response. After knocking down FTSJ3, there was a decrease in IFN expression. Subsequently, upon MDA5 knockdown, IFN expression increased. This result confirms that the methyltransferase contributes to the escape of HIV RNA 2'-O-methylation from MDA5 sensing, thus completing immune evasion.
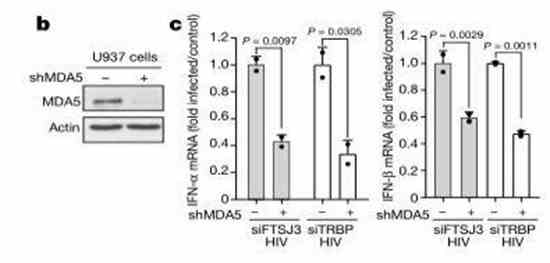 Figure 5: FTSJ3 Modulates MDA5-IFN Pathway Sensitivity.
Figure 5: FTSJ3 Modulates MDA5-IFN Pathway Sensitivity.
Depletion of FTSJ3 Suppresses HIV Replication:
To assess the impact of FTSJ3 on cellular immune stimulation by the virus, the authors employed FTSJ3 knockout in MDDCs during viral infection. Clearly, FTSJ3 knockout cells induced the expression of type I antiviral response to HIV virus particles, concurrently inhibiting the efficiency of HIV-1 expression. Thus, siFTSJ3-HIV non-methylated RNA induces innate immunity against viral RNA, suppressing HIV replication.
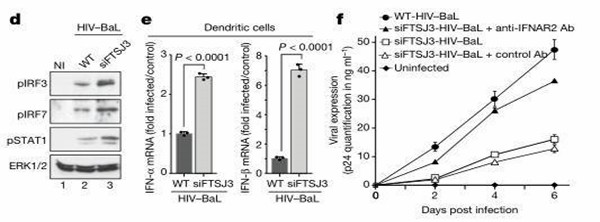 Figure 6: Depletion of FTSJ3 Suppresses HIV Replication.
Figure 6: Depletion of FTSJ3 Suppresses HIV Replication.
In summary, this study unveils, for the first time, the RNA 2'O-methylation activity of FTSJ3 and confirms that HIV virus can recruit the FTSJ3-TRBP system post-infection to accomplish 2'O-methylation modifications on viral RNA. Consequently, this modification lowers the sensitivity of the MDA5-IFN pathway, reducing immune system recognition. This novel pathway of HIV-1 innate immune evasion holds promise as a new therapeutic target for AIDS treatment.
References
- Dai Q, Moshitch-Moshkovitz S, Han D, et al. Nm-seq maps 2'-O-methylation sites in human mRNA with base precision. Nature methods, 2017, 14(7): 695-698.
- Ringeard, M., Marchand, V., Decroly, E. et al. FTSJ3 is an RNA 2'-O-methyltransferase recruited by HIV to avoid innate immune sensing. Nature, 2019, 565, 500–504.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Restriction endonuclease cleavage of phage DNA enables resuscitation from Cas13-induced bacterial dormancy
Journal: Nature microbiology
Year: 2023
IL-4 drives exhaustion of CD8+ CART cells
Journal: Nature Communications
Year: 2024
High-Fat Diets Fed during Pregnancy Cause Changes to Pancreatic Tissue DNA Methylation and Protein Expression in the Offspring: A Multi-Omics Approach
Journal: International Journal of Molecular Sciences
Year: 2024
KMT2A associates with PHF5A-PHF14-HMG20A-RAI1 subcomplex in pancreatic cancer stem cells and epigenetically regulates their characteristics
Journal: Nature communications
Year: 2023
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch
Journal: Science Advances
Year: 2024
Genomic imprinting-like monoallelic paternal expression determines sex of channel catfish
Journal: Science Advances
Year: 2022
See more articles published by our clients.


