
 Sample Submission Guidelines
Sample Submission Guidelines
EM-seq Service
DNA methylation is pivotal in regulating gene expression, influencing a wide array of biological processes including development, aging, and various diseases like cancer. As genomics research progresses, the demand for more efficient and accurate DNA methylation detection methods intensifies. EM-seq (enzymatic methylation sequencing) emerges as a novel DNA methylation analysis technology, taking the lead in precise methylation analysis with cutting-edge advantages. It offers unprecedented solutions with heightened sensitivity, accuracy, and minimal DNA input requirements.
At CD Genomics, we harness the power of EM-seq technology to deliver exceptional methylation analysis services, enabling you to explore the intricacies of gene regulation and the mechanisms underlying various diseases. In this comprehensive service page, we will delve into the key aspects of EM-seq, its technological advantages, application areas, and how we provide advanced support tailored to your research needs.
Introduction to EM-seq Technology
EM-seq is an enzyme-based DNA methylation sequencing technology that distinguishes between unmethylated cytosine © and methylated 5-methylcytosine (5mC) using enzymatic methods. Unlike the traditional bisulfite conversion method (WGBS), EM-seq employs enzymatic catalysis to convert unmethylated cytosines to uracil (U), while keeping methylated cytosines unaltered. During subsequent amplification and sequencing, uracil is recognized as thymine (T). This approach allows for precise detection of methylation sites.
Comparison of Enzymatic Conversion and Bisulfite Conversion
The conventional bisulfite conversion method is effective for detecting methylation. However, it requires harsh reaction conditions that often damage DNA, limiting its application in low-input DNA samples. EM-seq overcomes these issues by preserving DNA integrity, enabling it to process a broader range of samples, including trace and fragile DNA samples such as circulating tumor DNA (ctDNA).
Experimental Principle
EM-seq offers a comprehensive and robust solution for identifying methylation regions in the human genome. During library preparation, a unique enzymatic conversion is employed, causing much less damage to DNA and requiring smaller sample inputs, resulting in higher quality and better-performing libraries. The Twist custom methylation probe design provides effective, specialized probes for targeted CpG enrichment. Optimized hybridization reagents add flexibility to workflow timings and enhance on-target rates.
Methylation sequencing involves enzyme or chemical methods that convert unmethylated cytosine to uracil through deamination, while methylated cytosine remains intact. During amplification, complementary adenine pairs with uracil on the complementary strand, introducing thymine at the original unmethylated cytosine position. The sequence end product is asymmetrical, producing two different double-stranded DNA molecules post-conversion. For methylated DNA, the process results in an alternate sequence pattern, illustrating the distinction between methylated and unmethylated regions in DNA sequences.
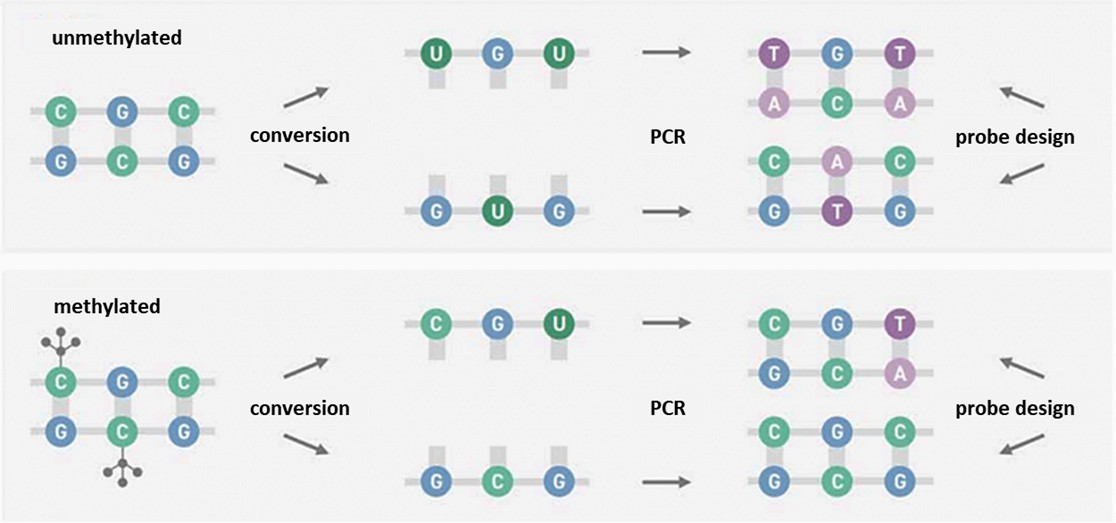 Figure 1. Methylation sequencing involves enzymatic or chemical methods.
Figure 1. Methylation sequencing involves enzymatic or chemical methods.
In the initial phase of the reaction, Ten-Eleven Translocation Dioxygenase 2 (TET2) plays a crucial role in transforming methylated cytosines such as 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) into 5-carboxycytosine (5caC). This is further enhanced by an oxidation booster, 5-glucosylhydroxymethylcytosine (5ghmC). These reactions serve as protective mechanisms, shielding 5mC and 5hmC from subsequent deamination activities.
Before deamination of cytosines to uracil by the enzyme APOBEC, the DNA undergoes denaturation to prepare for further reactions. Subsequent polymerase chain reaction (PCR) amplification transforms modified 5mC or 5hmC into cytosine and converts uracil into thymine. Post-PCR, the nucleic acid sequence mirrors that of bisulfite-converted sequences, ensuring compatibility of EM-seq with existing analytical workflows including tools like Bismark and bwa-meth.
Efficiency of Enzymatic Conversion
| Metric | Unmethylated Lambda DNA | CpG Methylated pUC19 DNA |
|---|---|---|
| Expected Conversion Efficiency | ≥99.5% | ≥99.5% |
| Measured Conversion Efficiency | 99.77% | 99.57% |
| Expected CpG Methylation Level | ~0.5% | 95-98% |
| Measured CpG Methylation Level | 0.22228% | 95.7572% |
Advantages of EM-seq
EM-seq surpasses the traditional bisulfite conversion method in several key aspects, particularly when working with trace amounts of DNA. Here are some primary advantages of EM-seq technology:
1. Low DNA Input and High Sensitivity: One of the major benefits of EM-seq is its ability to obtain high-quality sequencing data from minimal DNA quantities. Unlike WGBS, EM-seq causes less damage to DNA, allowing sequencing with as little as 10 ng of DNA. This feature is invaluable for analyzing rare or precious samples, such as circulating tumor DNA (ctDNA) in plasma, which are typically low in concentration and molecular weight.
2. Longer, More Complete Library Fragments: Compared to WGBS, EM-seq generates longer and more intact DNA fragments, minimizing sequencing gaps. This increases the comprehensiveness of genome-wide methylation information, particularly in regions that are difficult to sequence, by effectively reducing sequencing blind spots caused by short DNA fragments.
3. Superior GC Coverage Uniformity: DNA regions rich in GC content often pose challenges in sequencing due to potential biases that can affect accuracy. EM-seq excels in this area, ensuring uniform coverage across both GC-rich and AT-rich areas, thereby avoiding the GC coverage bias common in WGBS. This characteristic is crucial for accurately analyzing methylation states across the entire genome.
4. More Accurate CpG Island Detection: EM-seq is particularly effective in detecting CpG islands. At equivalent sequencing depths, EM-seq can uncover more CpG sites, many of which might be missed by traditional WGBS methods. This capability is particularly significant for early screening of diseases such as cancer, where methylation changes in CpG islands can serve as early disease markers.
Applications of EM-seq
In today's rapidly advancing scientific landscape, the EM-seq technology has emerged as a powerful tool with extensive applications across various fields. Its adeptness at detecting low concentration DNA samples and studying shifts in DNA methylation makes it particularly valuable. Here's how EM-seq is transforming key areas of research and development:
- Unveiling Cancer Early: Screening and Liquid Biopsies: EM-seq plays a crucial role in the early detection and ongoing monitoring of diseases by identifying cfDNA methylation markers in bodily fluids like plasma and urine.
- Diving into Epigenetics: This technique aids in uncovering how DNA methylation influences gene expression and biological processes, shedding light on the epigenetic variations across different biological states.
- Precision in Trace Sample Analysis: Whether it's precious samples such as oocytes, spermatocytes, or embryos, EM-seq delivers high-accuracy methylation insights, even from minuscule samples.
- Pioneering Biomarker Discovery: EM-seq is instrumental in the selection and validation of methylation markers related to specific biological processes, supporting future research and development efforts.
- Genomic and Epigenetic Insights into Diseases: By deciphering DNA methylation's role, EM-seq contributes significantly to our understanding of biological mechanisms, providing insights into complex disease processes.
- Enhancing Drug Development: The technology is leveraged to study how drugs affect DNA methylation patterns, offering critical molecular data that facilitate the screening and optimization of pharmaceuticals.
- Exploring Biodiversity and Evolution: EM-seq analyzes methylation patterns across different species or populations, helping to uncover adaptive changes and the genetic mechanisms governing them.
- Analyzing Transcription Start Site Methylation: It accurately assesses the methylation status at transcription start sites (TSS), particularly excelling in GC-rich regions, to reveal their regulatory roles in gene expression.
Platform Comparison for DNA Methylation Analysis
| Project Category | Platform | Features | Data Volume | Loci (10X) | Initial Amount (μg) | Technical Principle | Sample Requirement (Recommended) |
|---|---|---|---|---|---|---|---|
| 850K | Chip | Single-base resolution | / | 860,000 | 0.25 | Bisulfite Conversion | Multiples of 8 |
| WGBS | High-throughput | Single-base resolution | 90G | 5 million | 1 | Bisulfite Conversion | / |
| MC-seq | High-throughput | Single-base resolution | 20G | 2.7 million (84M) | 1 | Bisulfite Conversion | / |
| EM-seq | High-throughput | Single-base resolution | 25G | 4 million (134M) | 0.01 | Enzymatic | Multiples of 8 |
| scWGBS | High-throughput | Single-base resolution | 15G | 5 million | 0.01 | Bisulfite Conversion | / |
| Pyrosequencing | First-gen | Single-base resolution | 50 - 90 bp | / | 0.5 | Bisulfite Conversion | / |
EM-seq Workflow
Engaging with our services provides a seamless experience tailored to deliver high-quality results for your EM-seq projects. Here's how we ensure excellence at every step:
Step 1: DNA Extraction: We begin by meticulously extracting high-quality genomic DNA from your samples. This critical first step ensures that the DNA is intact and free from any contaminants, establishing a solid foundation for further analysis.
Step 2: Library Preparation: Using cutting-edge library preparation technologies, we adeptly prepare your DNA samples for EM-seq sequencing. This process is crucial for ensuring that your samples are optimally primed for accurate results.
Step 3: Sequencing: Your DNA library is then sequenced using the latest next-generation sequencing (NGS) technologies. This phase allows us to capture comprehensive genome-wide methylation data, providing an expansive view of the epigenetic landscape.
Step 4: Data Analysis: Our expert bioinformatics team crafts a detailed data analysis report for you. This report encompasses methylation levels, identification of differentially methylated regions, and detection of CpG islands, enabling you to glean actionable insights from your data.
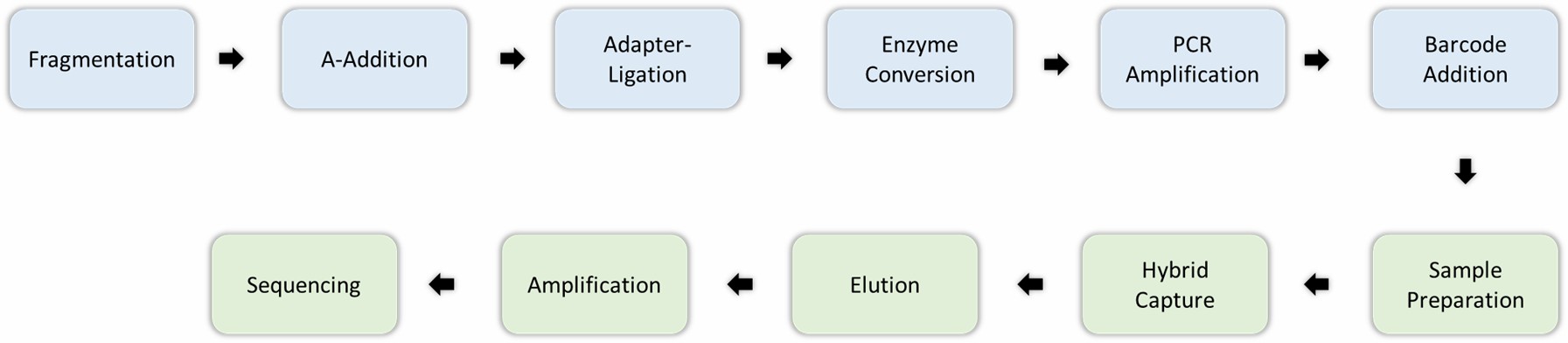 Figure 1. Methylation sequencing involves enzymatic or chemical methods.
Figure 1. Methylation sequencing involves enzymatic or chemical methods.
Service Specification
Sample Requirements
|
|
 Click |
Sequencing Strategies
|
 |
Bioinformatics Analysis We provide multiple customized bioinformatics analyses: 1. Genome Alignment Statistics 2. Overall Methylation Level Assessment
|
Analysis Pipeline
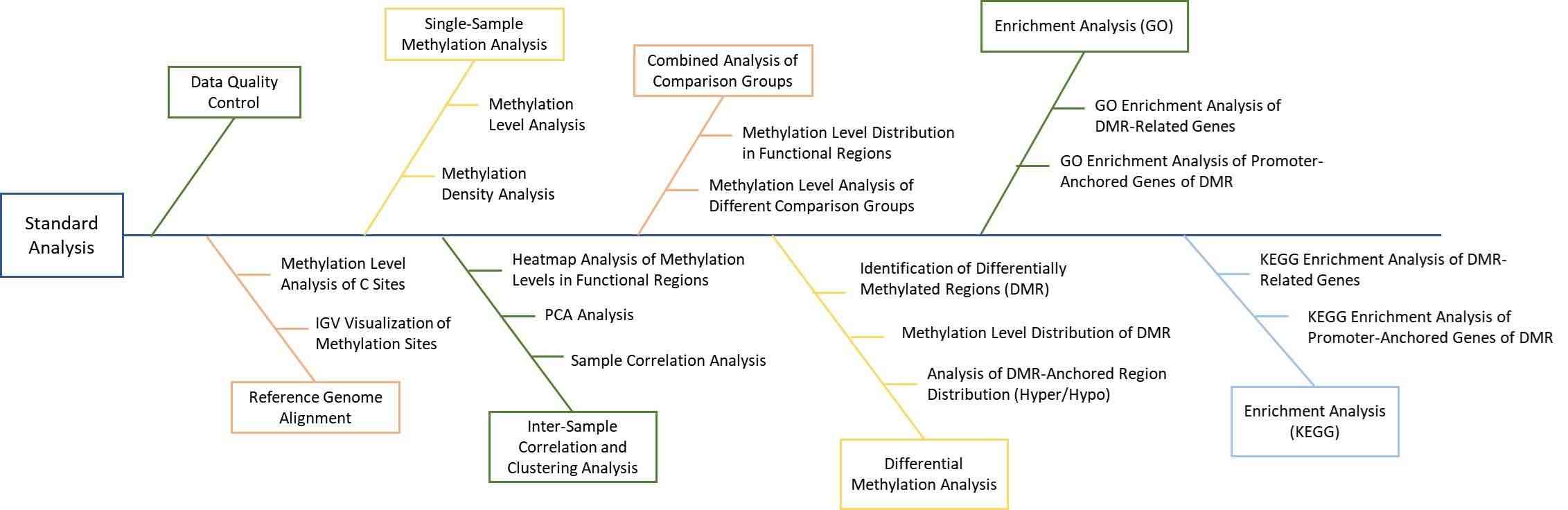
Deliverables
- Original Sequencing Data
- Experimental Results
- Data Analysis Report
- Detailed Methylation Profiling
EM-seq, as a revolutionary DNA methylation sequencing technology, surpasses the traditional bisulfite conversion method with its advantages in low DNA input, high sensitivity, and uniform GC coverage, providing higher-quality and more accurate methylation data for various research fields. At CD Genomics, we are committed to providing our customers with the most advanced EM-seq services to assist you in in-depth exploration of gene regulation, disease mechanisms, cancer research, and other areas.
Whether you are conducting basic research or developing advanced diagnostic tools, EM-seq services can provide excellent support for your research. Feel free to contact us to learn more about EM-seq or request a quote, and let this advanced technology help you achieve new scientific breakthroughs.
Demo Results
Partial results are shown below:
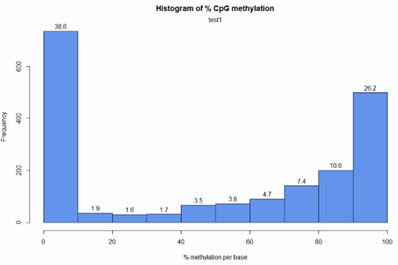
CpG Methylation Distribution Histogram
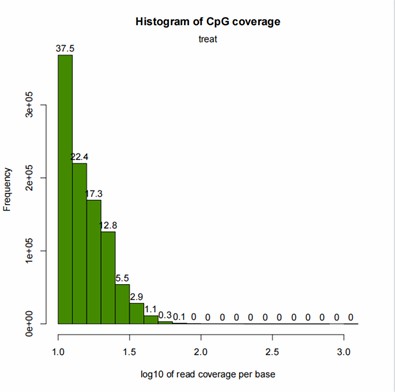
CpG Coverage Histogram
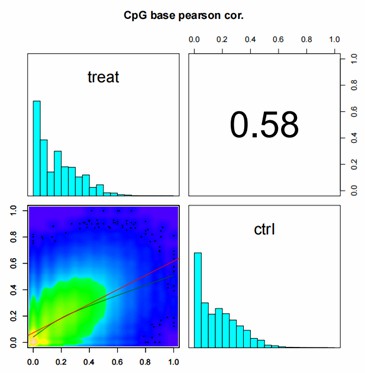
Correlation Analysis Between Samples
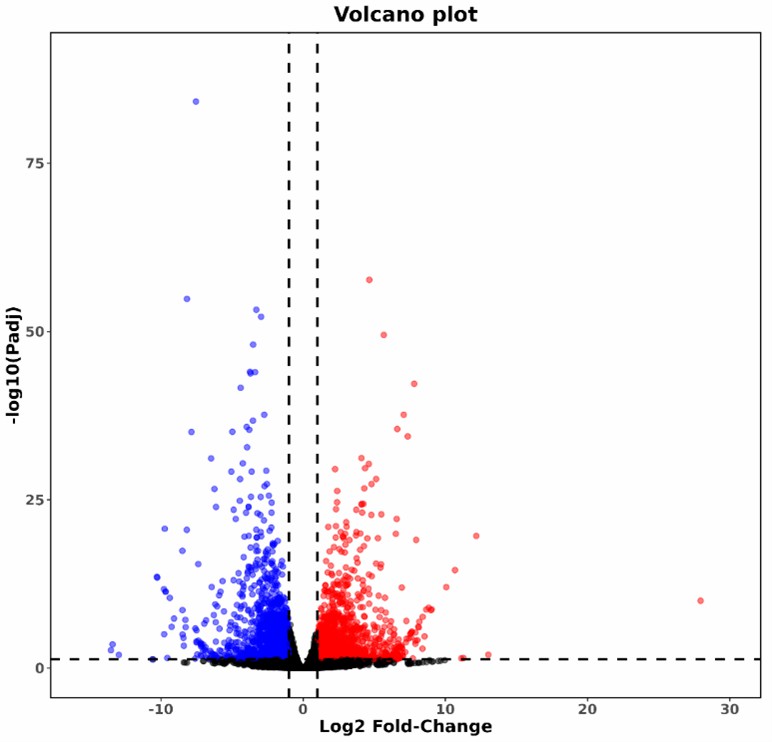
Volcano Plot of Differentially Methylated Sites
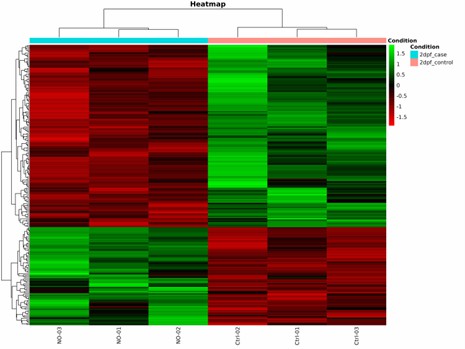
Heatmap of Differentially Methylated Sites
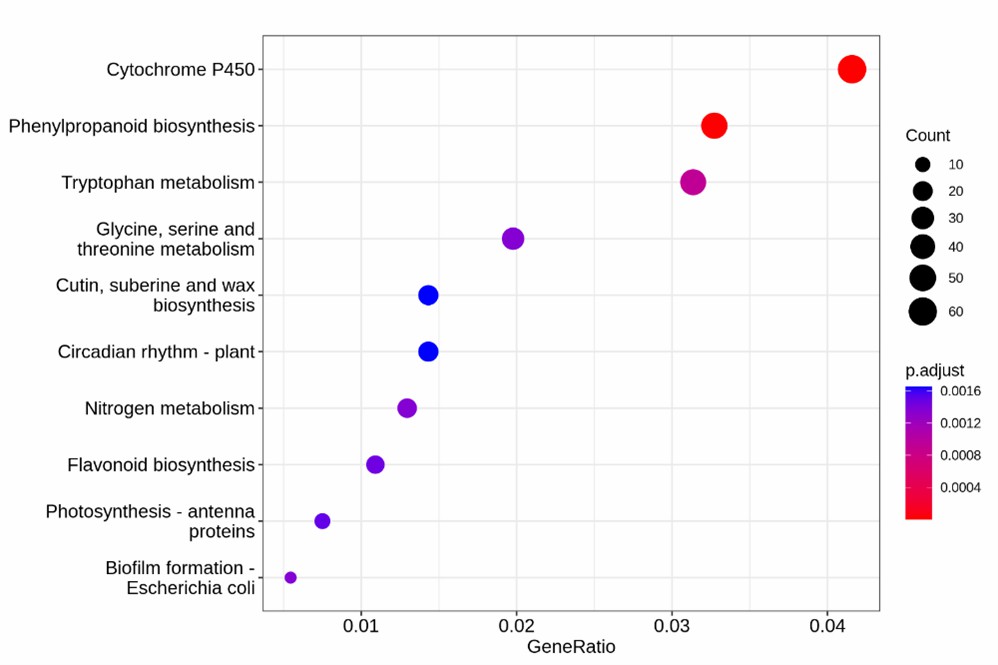
KEGG Enrichment Analysis of Differentially Methylated Regions
EM-seq FAQs
What should be noted when extracting gDNA or cfDNA?
When extracting genomic DNA (gDNA) or cell-free DNA (cfDNA), it is recommended to achieve the following:
- gDNA Requirement: Total ≥20 ng, Concentration ≥2 ng/μl
- cfDNA Requirement: Total ≥20 ng, Concentration ≥2 ng/μl
Additionally, it is crucial to avoid using EDTA or EB as solvents for your extracted gDNA/cfDNA, as these components can adversely affect enzymatic efficiency.
What precautions should be taken when separating serum or plasma?
When separating serum or plasma:
- Avoid Freeze-Thawing: Whole blood samples should not undergo freeze-thaw cycles.
- Timely Processing: Prepare plasma or serum as soon as possible after blood collection.
- Storage Conditions: Serum or plasma can be stored at -80°C to maintain integrity. Repeated freeze-thaw cycles should be avoided to preserve the sample quality.
How is sample conversion rate calculated?
During methylation library preparation, the theoretical conversion assumes all unmethylated cytosines © are converted to thymines (T) (originally converted to uracil (U), then to T during PCR). However, not all sites may convert, and the DNA's methylation status is initially unknown. Therefore, actual conversion rates can't be directly determined from the sample DNA.
To address this, lambda DNA (bacteriophage DNA), which contains only unmethylated cytosine, is used as a negative control with known methylation status. By incorporating lambda DNA in the library preparation, processing, and sequencing alongside the sample DNA, the conversion rate of lambda DNA is calculated to serve as an indicator of the sample's overall conversion rate.
EM-seq Case Studies
Multimodal Epigenetic Sequencing Analysis (MESA) of Cell-free DNA for Non-invasive Cancer Detection
Journal: Genome Medicine
Impact factor: 7.324
Published: 16 January 2024
Background
Cell-free DNA (cfDNA) methylation has emerged as a promising biomarker for early cancer detection, overcoming the limitations of genetic alteration-based approaches. Enzymatic Methyl-seq (EM-seq) improves upon traditional bisulfite sequencing by preserving DNA integrity, enabling multimodal epigenetic analysis that enhances cancer detection accuracy.
Materials & Methods
- Whole blood sample
- Clarified plasma
- Targeted sequencing
- Targeted EM-seq
- Data processing and quality control
- Multimodal feature extraction
- Feature selection
- Cross-cohort validation analysis
Results
MESA (Multimodal Epigenetic Sequencing Analysis) demonstrated strong performance in detecting colorectal cancer by integrating multiple epigenetic features from cfDNA. Key findings include:
1. Methylation Analysis:
- cfDNA methylation alone distinguished cancer from non-cancer samples with high accuracy (AUC = 0.8663 for cohort 1 and AUC = 0.8293 for cohort 2).
- Cancer samples showed higher methylation levels at target CpG sites, consistent with promoter hypermethylation in tumors.
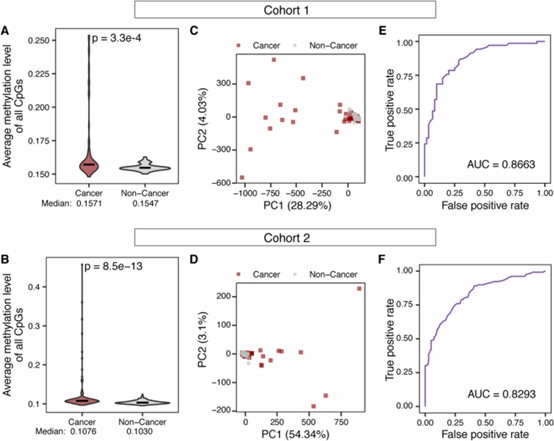 Differential cfDNA methylation between cancer and non-cancer samples enables accurate cancer detection.
Differential cfDNA methylation between cancer and non-cancer samples enables accurate cancer detection.
2. Nucleosome Organization Insights:
- MESA effectively captured nucleosome occupancy and fuzziness, revealing key structural differences between cancer and normal samples.
- Polyadenylation sites were included in the analysis for the first time, providing novel insights into cancer-related chromatin organization.
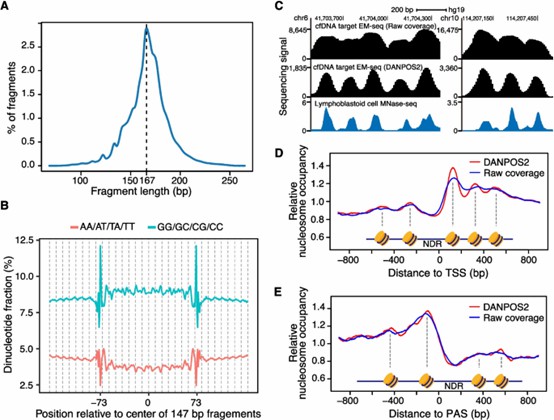 Nucleosome organization information from targeted EM-seq of cfDNA.
Nucleosome organization information from targeted EM-seq of cfDNA.
3. Cancer Detection Using Nucleosome Features:
- Nucleosome occupancy alone achieved AUCs of 0.8494 (cohort 1) and 0.9213 (cohort 2).
- Inclusion of polyadenylation site data further improved cancer detection accuracy.
- Nucleosome fuzziness, a novel metric capturing chromatin heterogeneity, provided additional predictive power.
4. Multimodal Integration for Enhanced Accuracy:
- Combining methylation, nucleosome occupancy, fuzziness, and WPS improved classification performance.
- The multimodal model consistently outperformed single-feature models across different cancer stages and cohorts.
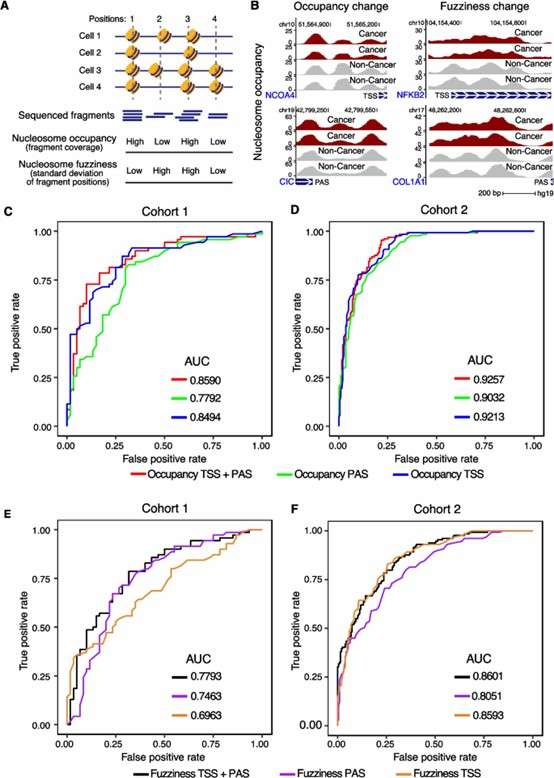 Accurate detection of cancer based on nucleosome occupancy and fuzziness.
Accurate detection of cancer based on nucleosome occupancy and fuzziness.
5. Cross-Cohort Validation:
- MESA was robust across different cohorts, maintaining high predictive accuracy.
- The approach was adaptable to other bisulfite-free sequencing methods (e.g., cfDNA TAPS) and was effective in detecting hepatocellular and pancreatic cancers.
Conclusion
MESA integrates multiple epigenetic modalities, enhancing detection accuracy for colorectal, liver, and pancreatic cancers, validated across four cohorts. It represents a major breakthrough in non-invasive cancer detection by leveraging comprehensive cfDNA epigenetic profiles.
Reference:
- Li, Y., Xu, J., Chen, C. et al. Multimodal epigenetic sequencing analysis (MESA) of cell-free DNA for non-invasive colorectal cancer detection. Genome Med 16, 9 (2024). https://doi.org/10.1186/s13073-023-01280-6
Related Publications
Here are some publications that have been successfully published using our services or other methylation sequencing services:
Genomic imprinting-like monoallelic paternal expression determines sex of channel catfish
Journal: Science Advances
Year: 2022
Folate Carrier Deficiency Drives Differential Methylation and Enhanced Cellular Potency in the Neural Plate Border
Journal: Frontiers in Cell and Developmental Biology
Year: 2022
Temporal genome-wide DNA methylation signature of post-smolt Pacific salmon challenged with Piscirickettsia salmonis
Journal: Epigenetics
Year: 2021
KMT2A associates with PHF5A-PHF14-HMG20A-RAI1 subcomplex in pancreatic cancer stem cells and epigenetically regulates their characteristics
Journal: Nature communications
Year: 2023
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch
Journal: Science Advances
Year: 2024
Genomic imprinting-like monoallelic paternal expression determines sex of channel catfish
Journal: Science Advances
Year: 2022
See more articles published by our clients.


