
 Sample Submission Guidelines
Sample Submission Guidelines
Pre-made Library Sequencing
The Introduction of Pre-made Library Sequencing
CD Genomics accepts customers' prepared libraries for sequencing. You can submit the libraries for full QC test and use our sequencing-only service. We provide the most advanced and powerful sequencing platforms with different capacities and read lengths to fit any project scale, budget, and turnaround. Our QC library ensures optimal cluster generation and maximal data output for each run. Different samples can be multiplexed and sequenced together as long as they have the same read length. Our highly experienced team offers consultation on the best sequencers for different research goals. We also offer FREE PhiX spike-in, demultiplexing (FASTQ), and data transfer via FTP.
Prefabricated library sequencing is a high-throughput sequencing technique utilized for the systematic analysis of the genome, transcriptome, or other nucleic acid sequences of biological samples. It involves preparing nucleic acids from the sample into libraries, followed by sequencing using high-throughput sequencers, thereby generating a vast amount of sequence data for further analysis. Prefabricated libraries entail fragmenting the sample's nucleic acids before sequencing, and through a series of biochemical reactions, incorporating adapter sequences compatible with the sequencing platform, rendering them suitable for high-throughput sequencers.
If you want to learn more about pre-made library sequencing, you can refer to our article "The Introduction and Workflow of Pre-made Library Sequencing."
Advantages of Pre-made Library Sequencing
- High-Throughput: Capable of generating a large amount of sequence data in a short timeframe.
- Versatility: Applicable to various types of nucleic acid samples (DNA, RNA, etc.).
- Flexibility: Enables research at different levels such as genomics, transcriptomics, epigenomics, etc.
- Accuracy: High-throughput sequencing technology exhibits high accuracy and coverage.
- Reliable Expertise: Proficient team expertise guides you through every step of sample processing.
- Quick Turnaround Time: Provides the fastest turnaround time for sample processing, allowing quicker progression from quality control to data dissemination and expediting project timelines.
Application of Pre-made Library Sequencing
- Genomics Research
- Transcriptomics Research
- Epigenetics Research
- Microbiomics Research
- Oncology Research
- Rare Diseases Research
- Agricultural and Breeding Research
- … and more
Pre-made Library Sequencing Workflow
The quality inspection method of the sizes and concentrations of the library is Qubit, Agilent bioanalyzer.
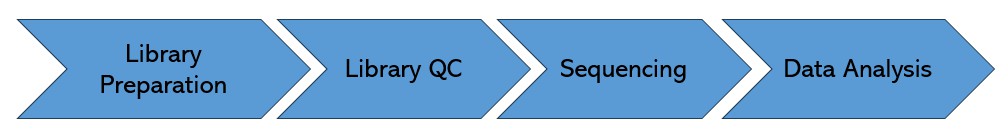
Service Specifications
Sample Requirements
| Platform | Minimum Concentration | Data Amount | Volume Requirement |
|---|---|---|---|
| Novaseq-PE150 | 2 ng/μL | X<30G 30G≤X<100G 100G≤X≤400G 400G<X<800G 800G |
≥15 μL ≥25 μL ≥50 μL ≥70 μL ≥100 μL (additional 70μL for one more lane) |
| Nova- PE250 | 2 ng/μL | X<30GM 30M≤X<100M 100M≤X<400M 400M |
≥15 μL ≥25 μL ≥50 μL ≥100 μL (additional 70μL for one more lane) |
| HiSeq-PE150 | 1 ng/μL | 1 Lane | ≥10 μL |
| MiSeq-PE300 | 1 ng/μL | 1 Flowcell | ≥10 μL |
Note: Sample amounts are listed for reference only. For detailed information, please contact us with your customized requests.
Sequencing Strategy
Illumina Sequencing:
| Platform | Read Length (nt) | Unit | Unit Output (raw clusters in Millions) |
|---|---|---|---|
| NovaSeq 6000 Click |
PE50 | Lane (SP) | 375-400 M |
| PE50 | Lane (S1) | 650-800M | |
| PE50 | Lane (S2) | 1,650-2,050 M | |
| PE100 | Lane (SP) | 375-400 M | |
| PE100 | Lane (S1) | 650-800M | |
| PE100 | Lane (S2) | 1,650-2,050 M | |
| PE100 | Lane (S4) | 4,000-5,000M | |
| PE150 | Lane (SP) | 375-400 M | |
| PE150 | Lane (S1) | 650-800M | |
| PE150 | Lane (S2) | 1,650-2,050 M | |
| PE150 | Lane (S4) | 4,000-5,0000M | |
| PE250 | Lane (SP) | 375-400 M | |
| HiSeq 4000 | SE50 | Lane | 300-400 M |
| PE75 | Lane | 300-400 M | |
| PE150 | Lane | 300-400 M | |
| MiSeq | PE150 | Flowcell (Nano) | 1 M |
| PE250 | Flowcell (Nano) | 1 M | |
| PE150 | Flowcell (Micro) | 4 M | |
| SE36 | Flowcell (V2) | 12-15 M | |
| PE25 | Flowcell (V2) | 12-15 M | |
| PE150 | Flowcell (V2) | 12-15 M | |
| PE250 | Flowcell (V2) | 12-15 M | |
| PE75 | Flowcell (V3i) | 22-25 M | |
| PE300 | Flowcell (V3) | 22-25 M |
| Platform | Run Type |
|---|---|
| Pacbio Sequl II SMRT Cell 8M | HiFi Sequencing |
| CLR Sequencing |
Bioinformatics Analysis
- Data Quality Control
- Data Preprocessing
- Alignment
- Assembly
- Variant Detection
- Gene Expression Analysis
- Functional Annotation
- … and more
Note: Recommended data outputs and analysis contents displayed are for reference only. For detailed information, please contact us with your customized requests.
Analysis Pipeline
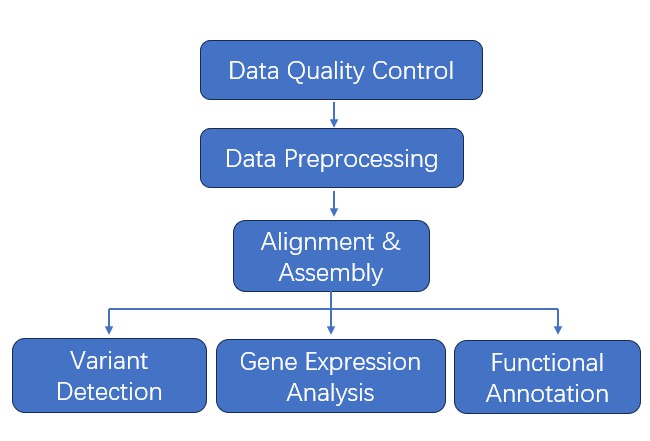
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Pre-made Library Sequencing for your writing (customization)
Demo Results
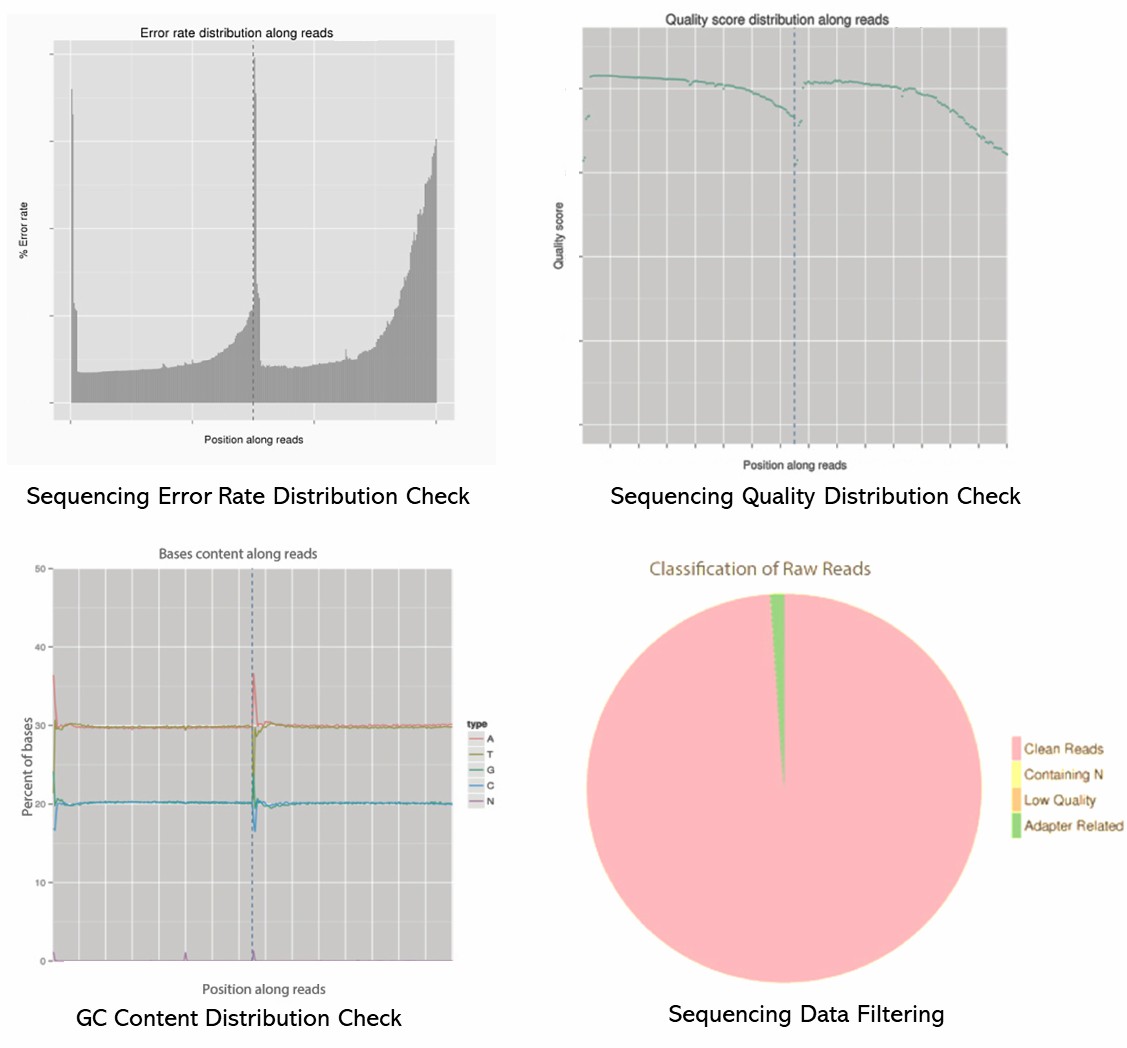
Pre-made Library Seq FAQs
1. What types of samples can be used for pre-made library sequencing?
For pre-made library sequencing, various sample types are employed, encompassing:
- Genomic DNA: Utilized for whole-genome sequencing (WGS) or targeted sequencing applications.
- RNA: Employed in transcriptome sequencing (RNA-seq), encompassing total RNA, mRNA, and small RNA.
- cDNA: Employed for sequencing reverse-transcribed RNA in specific contexts.
2. How is the quality of the pre-made library assessed?
The quality of the pre-made library is assessed through:
- Size Distribution: Analyzing the fragment size distribution using an instrument like the Agilent Bioanalyzer or TapeStation.
- Concentration Measurement: Quantifying the library concentration using a Qubit fluorometer or quantitative PCR (qPCR).
- Quality Control Metrics: Checking for adapter dimers, contamination, and overall quality using tools like FastQC for raw sequencing data.
3. What does the uniformity of library output refer to?
During sequencing, the output data volume of libraries in the same lane remains uniform relative to the amount of library input. For example, when mixing 5 libraries and obtaining a total of 50G data, there should not be a scenario where one library yields 30G of data while another only yields 5G; each library should ideally contribute equally at 10G.
4. Can pre-made library sequencing be utilized for single-cell analysis?
Certainly, pre-made library sequencing can be repurposed for single-cell analysis. Techniques like single-cell RNA sequencing (scRNA-seq) entail isolating individual cells, reverse transcribing RNA into cDNA, and creating sequencing libraries from the resulting single-cell cDNA. This methodology enables the investigation of gene expression at the single-cell level, unraveling insights into cellular heterogeneity and function.
Pre-made Library Seq Case Studies
Landscape of somatic mutations in 560 breast cancer whole genome sequences
Journal: Nature
Impact factor: 64.8001
Published: 2 May 2016
Background
The mutational theory of cancer postulates that specific DNA sequence alterations, referred to as "driver" mutations, confer proliferative advantages to cells, fostering the emergence of malignant cell populations. These mutations can stem from diverse mechanisms, encompassing exposure to mutagens, errors in DNA repair mechanisms, and inaccuracies during DNA replication. Technological progress, spanning from karyotype analysis to high-throughput DNA sequencing, has substantially enhanced the delineation of cancer-associated mutations. Nonetheless, predominant attention has been directed towards protein-coding regions, leaving crucial inquiries unanswered regarding mutations within non-coding regions and the intrinsic mutagenic processes underlying breast cancer. To bridge these knowledge gaps, we conducted an in-depth analysis of whole-genome sequences from 560 breast cancer cases, striving for a comprehensive elucidation of somatic mutations in this context.
Methods
- 560 breast cancers
- Normal tissue
- DNA extraction
- Total RNA extraction
- Short insert 500bp genomic libraries
- 350bp poly-A selected transcriptomic libraries
- High-throughput sequencing
- Alignment
- Processing of genomic data
- Identification of novel breast cancer genes
- Mutational signatures analysis
- Rearrangement signatures
- Individual patient whole genome profiles
Results
Whole genomes of 560 breast cancers and matched non-neoplastic tissues were sequenced, detecting numerous somatic mutations. By combining data from various sources, new cancer genes were identified, and driver mutations were defined. Genomic rearrangements and copy number changes further contributed to the identification of driver mutations, with TP53, PIK3CA, and MYC being among the most frequently mutated genes.
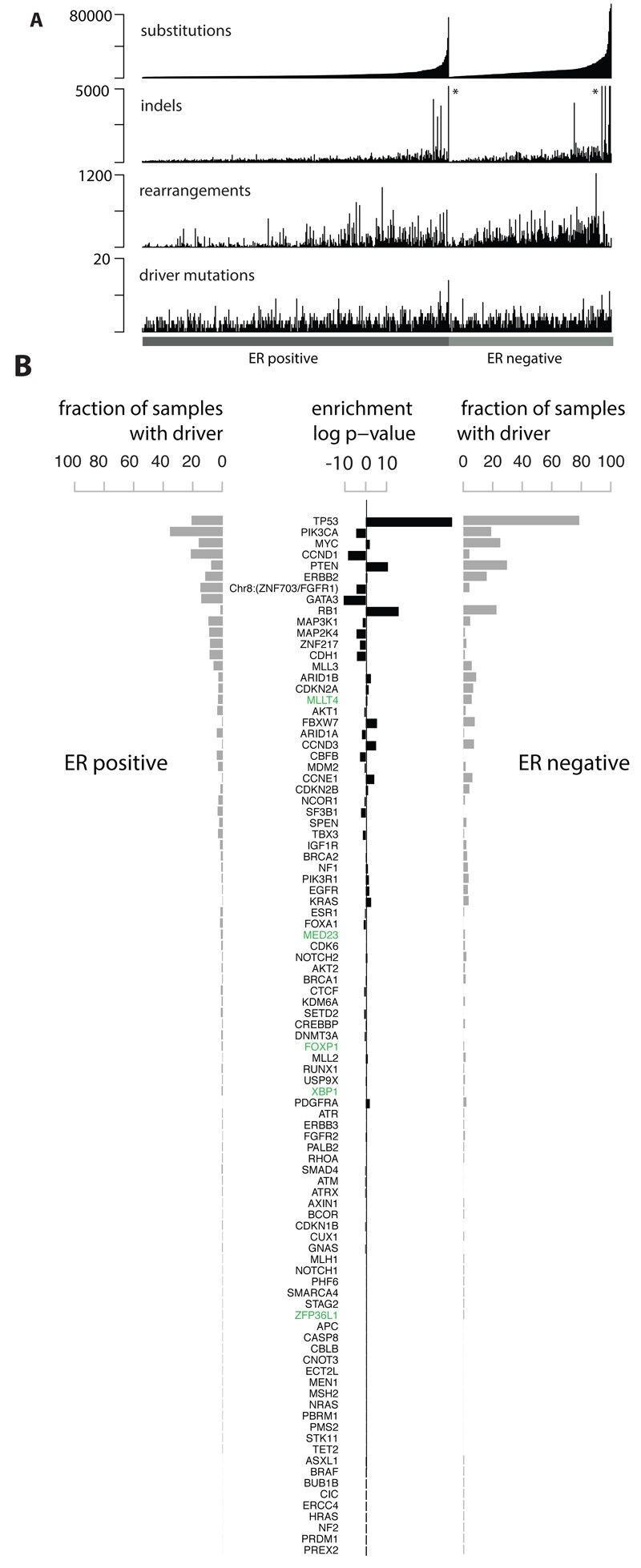 Fig 1. Cohort and catalogue of somatic mutations in 560 breast cancers.
Fig 1. Cohort and catalogue of somatic mutations in 560 breast cancers.
We explored non-coding somatic mutations and identified recurring mutations in the PLEKHS1 promoter linked to mutational signatures 2 and 13. Analogous mutations were noted in the promoters of TBC1D12 and WDR74, suggesting potential hypermutability. Additionally, mutations were detected in long non-coding RNAs MALAT1 and NEAT1, though their significance as driver mutations remains uncertain.
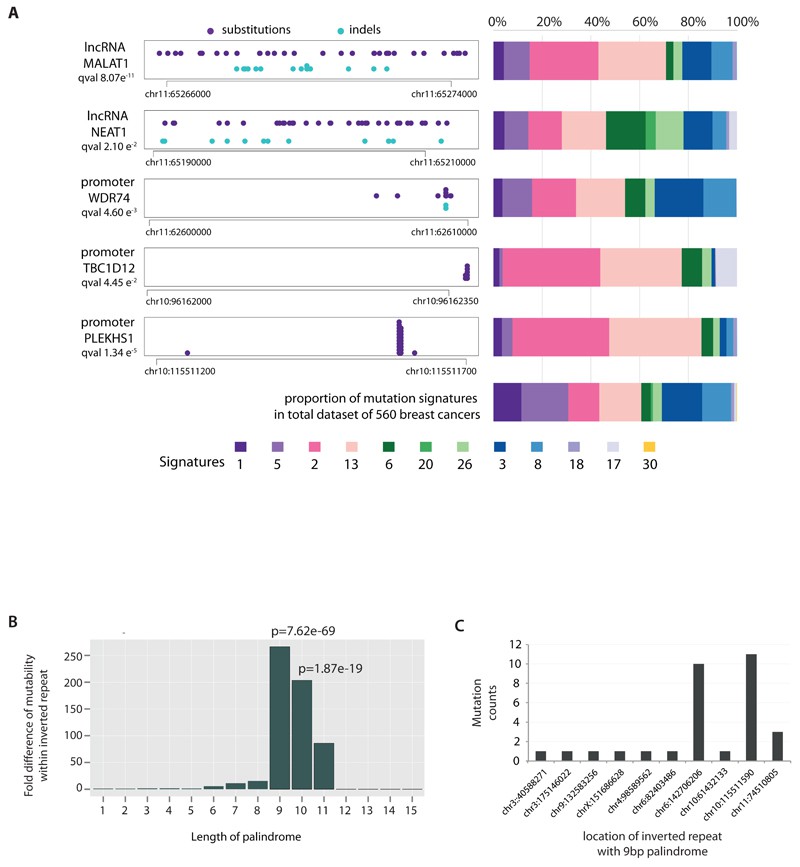 Fig 2. Non-coding analyses of breast cancer genomes.
Fig 2. Non-coding analyses of breast cancer genomes.
Conclusion
Progress towards a comprehensive understanding of breast cancer genetics is underway, revealing multiple mutational signatures and implicating numerous cancer genes. However, the rarity of dominantly-acting fusion genes and non-coding driver mutations suggests additional complexities. Yet, many questions remain, including the identification of additional cancer genes and the roles of viruses or microbes. Further exploration of whole genome sequences from breast cancer patients is needed to fully elucidate the somatic mutational landscape of the disease.
Reference:
- Nik-Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature, 2016, 534(7605): 47-54.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
The HLA class I immunopeptidomes of AAV capsid proteins
Journal: Frontiers in Immunology
Year: 2023
Isolation and characterization of new human carrier peptides from two important vaccine immunogens
Journal: Vaccine
Year: 2020
Change in Weight, BMI, and Body Composition in a Population-Based Intervention Versus Genetic-Based Intervention: The NOW Trial
Journal: Obesity
Year: 2020
Sarecycline inhibits protein translation in Cutibacterium acnes 70S ribosome using a two-site mechanism
Journal: Nucleic Acids Research
Year: 2023
Identification of a Gut Commensal That Compromises the Blood Pressure-Lowering Effect of Ester Angiotensin-Converting Enzyme Inhibitors
Journal: Hypertension
Year: 2022
A Splice Variant in SLC16A8 Gene Leads to Lactate Transport Deficit in Human iPS Cell-Derived Retinal Pigment Epithelial Cells
Journal: Cells
Year: 2021
See more articles published by our clients.


