
 Sample Submission Guidelines
Sample Submission Guidelines
The Introduction and Workflow of Pre-made Library Sequencing
What is Pre-made Library Sequencing
For next-generation sequencing, fully automated sequencing runs at a lower cost per base and faster assay times are available with lately introduced high throughput and benchtop instruments. As a result, an important bottleneck has been detected in the complex and time-consuming library preparation process, which begins with isolated nucleic acids and ends with amplified and barcoded DNA with sequencing adapters. Library preparation protocols are usually multistep processes that require expensive reagents and a lot of hands-on time. To guarantee robustness and reproducibility, a strong emphasis on standardization will be required.
Prefabricated library sequencing is a common high-throughput sequencing approach utilized for analyzing genomic information within DNA samples. The primary focus of pre-library sequencing lies in the preparation of DNA libraries, which contain prepped DNA fragments ready for sequencing. These DNA fragments usually stem from various samples used in biological research. Pre-library sequencing can be performed using second-generation sequencing platforms as well as third-generation sequencing platforms like PacBio SMRT Sequencing and Nanopore Sequencing.
Services you may interested in
What is the Workflow of Pre-made Library Sequencing
Preparing Sequencing Libraries:
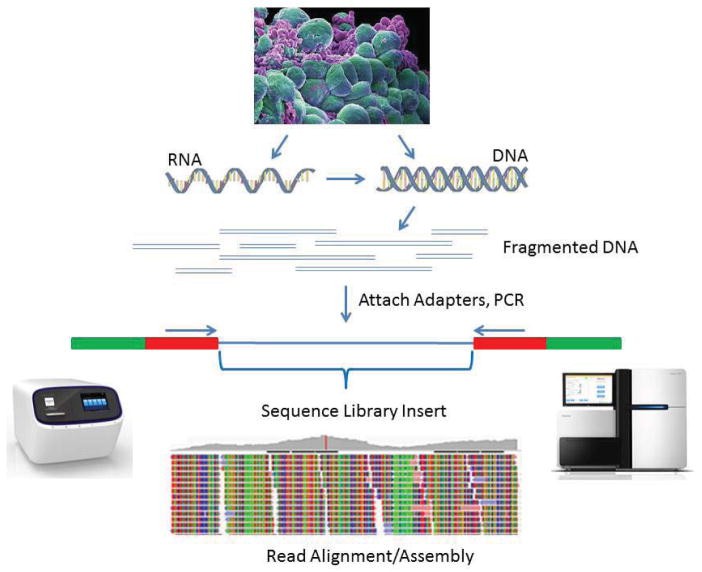 Figure 1. Basic workflow for NGS library preparation. (Head et al., 2014)
Figure 1. Basic workflow for NGS library preparation. (Head et al., 2014)
- DNA and RNA sequencing libraries
Starting with genomic DNA or RNA, a sequencing library can be created. The workflow for creating a DNA sequencing library is made up of three basic steps:
- Nucleic acid (DNA or RNA) fragmentation and sizing to acquire fragments of a predetermined length,
- Connection of adaptors (adapters) to the fragment extremities, and
- Library quantification.
There is an additional step in any RNA sequencing library: RNA conversion to cDNA. The fragmentation procedure can be performed either before or after the cDNA synthesis.
- Fragmentation
Physical methods, enzymatic methods, and chemical methods can all be used to fragment nucleic acids (DNA, RNA, or cDNA). The most frequently used techniques are physical and enzymatic. Specifically, long fragments can be achieved for mate-pair libraries (6,000 to 20,000 bp).
- Fragment sizing
The size of the fragments is extremely important. The ideal library fragment size is determined by the platform to be used and the scope of the analysis. On Illumina platforms, for example, fragments of up to 1,500 bp can be used in the case of exome sequencing, however, a maximum insert size of 200-250 bp is suggested. This is due to a human exon's average size of 200 base pairs.
- Attachment of the adapters
The so-called adapters must be affixed to both extremities of each fragment once the DNA or RNA fragmentation is complete. By definition, a sequencing library is a collection of DNA fragments with adapters connected Adapters are made to work with a specific sequencing platform, such as the flow-cell surface or beads. Following the attachment of the adapters, a sizing phase occurs, during which all fragments of undesirable size and all adapter dimers are removed Adapter dimers form when adapters self-ligate without a library insert sequence, and they're especially common when the initial DNA quantity is low. It is critical to remove adapter dimers from the library because they can significantly reduce sequencing yield by consuming valuable flow cell space. A clean-up with magnetic beads can effectively erase the dimers.
- Library quantification
The library quantification is a critical step that should be carried out using the most precise and practical method possible. PCR-based methods (digital PCR or quantitative PCR) are commonly used to quantify sequencing libraries.
- The final quality of the sequencing library
When creating a sequencing library, it's critical to aim for the highest level of complexity possible. In other words, it is critical that the final library captures as much of the original material's uniqueness as possible. Limiting the number of segmental duplications is the first step toward achieving this result. The shorter the fragments are, the more likely they are to be less specific and align at multiple loci in the reference sequence. As a result, the percentage of duplicate reads in the sequencing data can be used to determine library complexity.
Sequencing:
After undergoing quality control and amplification procedures, the DNA library is processed for high-throughput sequencing on a sequencer. The sequencing of pre-made libraries typically employs platforms such as Illumina, which have the capability of producing a substantial quantity of short sequence reads.
Data Analysis:
Upon the completion of library sequencing, the resultant sequence data is transferred to a computational system for bioinformatics analysis. The analytic process encompasses sequential activities including quality control of sequences, alignment of sequences, and identification of variants. During the quality control phase, the original sequence data undergoes filtration to exclude sequences of low quality. Subsequent to this, through alignment with a reference genome or genome assembly, the source and positional attributes of DNA sequences can be discerned. Ultimately, mechanisms like variant detection enable the identification of genetic modifications within the sample, such as single nucleotide polymorphisms (SNPs) or structural variations (SVs), thereby revealing the genetic information characteristic of the DNA sample.
Conclusion
Pre-made library sequencing plays a vital role in various genomic studies. It serves as a cornerstone in genome sequencing, facilitating rapid acquisition of high-quality whole-genome sequence data and advancing genomic research across humans, plants, animals, and other organisms. In transcriptome sequencing, pre-made library sequencing comprehensively decodes gene expression profiles, aiding researchers in understanding gene regulatory mechanisms under different conditions.
Furthermore, pre-made library sequencing has significant applications in epigenomics research. It is utilized to analyze epigenetic markers such as DNA methylation, histone modifications, unveiling the intricate network of gene expression regulation. In the realm of genotyping and variant detection, pre-made library sequencing excels by accurately detecting single nucleotide polymorphisms (SNPs) and structural variations (SVs), offering crucial data support for personalized medicine and disease research.
In conclusion, pre-made library sequencing stands out as a robust and time-efficient method for analyzing genomes across various biological specimens. Its simplified workflow, beginning with DNA library construction and culminating in bioinformatic analyses, enables swift and precise genomic characterization. By circumventing extensive sample preparation procedures, pre-made library sequencing diminishes experimental discrepancies and lessens resource consumption.
CD Genomics offers pre-made library sequencing services, utilizing sequencing platforms with varying capacities and read lengths to meet diverse research needs. Additionally, we provide an extensive array of sequencing services, including Genomics Sequencing, Transcriptome Sequencing, Epigenomics Sequencing, and Genotyping. Our team of skilled professionals is dedicated to delivering exceptional support and assistance, ensuring the success of your research endeavors.
References:
- Hess JF, Kohl TA, Kotrová M, et al. Library preparation for next generation sequencing: a review of automation strategies. Biotechnology advances. 2020 Jul 1;41.
- Head SR, Komori HK, LaMere SA, et al. Library construction for next-generation sequencing: overviews and challenges. Biotechniques. 2014 Feb;56(2).
- Robin JD, Ludlow AT, LaRanger R, et al. Comparison of DNA quantification methods for next generation sequencing. Scientific reports. 2016 Apr 6;6(1).


