
 Sample Submission Guidelines
Sample Submission Guidelines
Human Whole Genome PacBio SMRT Sequencing
CD Genomics has been providing an accurate and affordable human genome resequencing service for a couple of years. CD Genomics introduces previously hidden PacBio SMRT technology that has great application potential in human genome resequencing. The long single-molecule reads reveal structural variants and produce direct variant phasing information across haplotype blocks and methylation. This is very helpful to broaden the utility of precision medicine efforts to improve human health, and greatly promote the development of human single-gene diseases, complex diseases and tumor genomes.
The Introduction of Human Whole Genome PacBio SMRT Sequencing
Having a comprehensive map of human genomic variations holds paramount importance for an in-depth understanding of genetic traits and for bolstering accurate disease research. Although short-read sequencing can reliably detect small variations such as SNP (Single Nucleotide Polymorphisms) and InDels (Insertions and Deletions), it presents limited sensitivity in identifying CNVs (Copy Number Variations) and SVs (Structural Variations). In recent years, long-read sequencing has gained widespread usage in human genomic research. This technique effectively deciphers complex genomic structures, inclusive of highly repetitive gene regions and structural genome variations which offers a more comprehensive genomic perspective in the quest for disease-associated variations. The high accuracy of long-read sequencing allows it to discover rare variations that short-read sequencing might miss, thus providing more precise genetic variation information, an essential input for precision medicine and its foundational research.
Human Whole Genome Pacific Biosciences Single Molecule, Real-Time (PacBio SMRT) Sequencing is an advanced genomic analysis technology incorporating the proprietary SMRT sequencing approach developed by Pacific Biosciences. This technology stands out due to its capacity to generate extended read lengths, demonstrate high sequence accuracy, and offer direct detection of epigenetic modifications, thereby allowing a comprehensive and profound analysis of the human genome. Remarkably, PacBio SMRT Sequencing is capable of generating extraordinarily long reads that typically range from 10-15 kilobase pairs (kb), and in some cases even exceed this length. This feature proves especially beneficial for deciphering intricate genomic structures, such as regions characterized by a high degree of sequence repetition or structural variation.
CD Genomics now employs PacBio SMRT sequencing for conducting comprehensive analysis on human whole genomes. With the ability to perform whole genome sequencing across various individual and population level samples, followed by profound bioinformatics analysis at both levels, this method paves the way for the broad exploration of genomic variations. Such variations include Single Nucleotide Polymorphisms (SNPs), Insertions and Deletions (InDels), Copy Number Variations (CNVs) and Structural Variations (SVs). The intimate genomic insights derived from this direct and extensive exploration are indispensable in identifying both pathogenic and susceptibility genes, as well as in understanding the mechanisms underlying disease onset and inheritance patterns.
Advantages of Human Whole Genome PacBio SMRT Sequencing
- Long-reads. It is beneficial for variation information mining of the whole genome, accurate analysis of chromosomal structural variants (SVs) and fusion genes
- No PCR amplification. Effectively avoiding the amplification bias, and easily spanning regions with high GC content and high sequence repetition, to ensure the integrity and homogeneity of genome coverage.
- Directly detects epigenetic modifications by measuring kinetic variation during base incorporation
Application of Human Whole Genome PacBio SMRT Sequencing
1. SV detection
SVs represent genomic rearrangements (typically defined as longer than 50 bp), and SVs may play important roles in human disease, evolution and genetic diversity. Many inherited diseases and cancers have been associated with a large number of SVs in recent years. There has been tremendous progress in the detection of single nucleotide variants (SNVs) in the past quarter-century, but intermediate-sized (50 bp to 50 kb) structural variants (SV) remain a challenge with short-read DNA sequencing. The longest read length of the PacBio SMRT sequences is 40~70 K, which can easily cover high repetition and high heterozygosity areas, providing the possibility for SVs detection.
2. Haplotype Genotyping
A haplotype (haploid genotype) is a group of alleles in an organism that is inherited together from a single parent. The correct genotyping of haplotype gene is important; for example, the outcome of unrelated donor marrow transplantation is influenced by donor-recipient matching for HLA. PacBio long reads can span multiple single nucleotides and structural variants, which directly phases the variants into haplotypes.
3. DNA Methylation
DNA methylation is a process by which methyl groups are added to the DNA molecule. It is essential for normal development and is associated with a number of key processes including genomic imprinting, X-chromosome inactivation, repression of transposable elements, aging and carcinogenesis. There are many ways to detect DNA methylation, including whole genome bisulfite sequencing (WGBS), methylated DNA Immunoprecipitation Sequencing (MeDIP) and so on. But these methods are difficult to operate experimentally.
While PacBio platform can describe the direct detection of DNA methylation, without bisulfite conversion according to the difference in the fluorescence pulse signal interval. In addition, the long reads sequencing allows for more thorough regional CpG methylation assessment and increases the capacity for studying the relationship between phased single nucleotide variants and allele-specific CpG methylation.
Human Whole Genome PacBio SMRT Sequencing Workflow

Service Specification
Sample Requirements
|
|
 |
Sequencing Strategy
|
 |
Bioinformatics Analysis
CD Genomics provides statistical and bioinformatic data analysis services on:
|
Analysis Pipeline
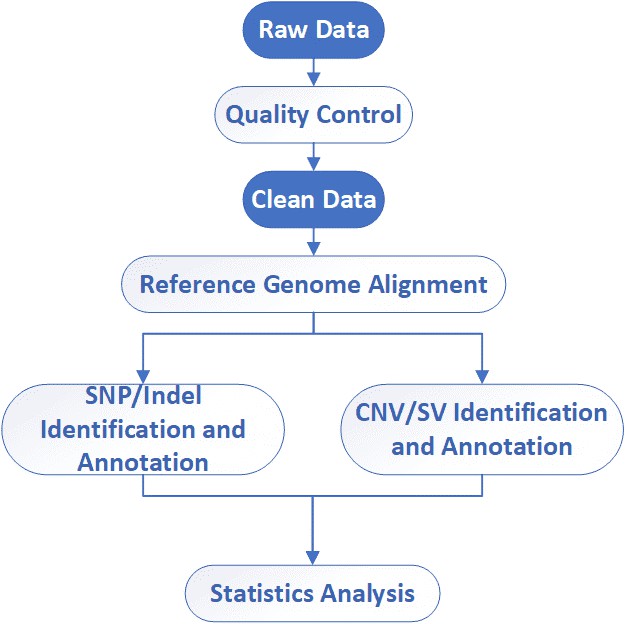
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Human Whole Genome PacBio SMRT Sequencing for your writing (customization)
CD Genomics provides full whole genome resequencing service package including sample standardization, library construction, deep sequencing, raw data quality control, and bioinformatics analysis. We can tailor this pipeline to your research interest. If you have additional requirements or questions, please feel free to contact us.
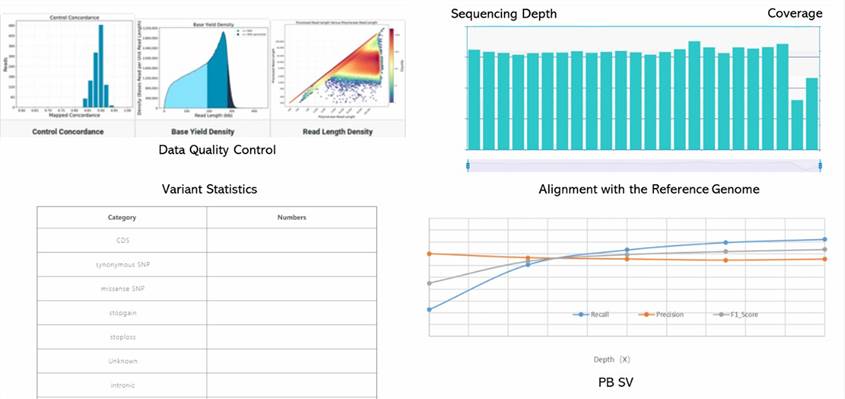
Demo Results
Human Whole Genome PacBio SMRT Seq FAQs
1. What is the depth of human whole-genome resequencing?
The sequencing depth is determined by research purpose, sample numbers, and your needs. The general depth of human whole-genome resequencing is 30X. For the detection of germline variations, we recommend sequencing depth of 30-50X, such as researches on single-gene disease. For population studies with multiple samples, if you focus on SNP, sequencing depth of 10X is enough. If you focus on structural variations in cancer tissues, we recommend sequencing depth should be more than 50X.
2. What methods can be used to validate the results?
Whole-genome resequencing can detect different types of genetic variations, including SNP, InDel, SV, and CNV.
- PCR amplification and sequencing or SNP genotyping can be used to validate SNPs.
- PCR amplification and Sanger sequencing can be used to validate short-fragment InDels.
- Real-time PCR are useful for validating CNVs.
- Small scale SVs can be validated by PCR amplification and sequencing, while large scale SVs need to be validated by microscopic observation, such as FISH.
3. What are the advantages of third-generation whole-genome sequencing over second-generation whole-genome sequencing?
In contrast to second-generation whole-genome sequencing, which utilizes library construction involving DNA fragments of approximately 350 base pairs (bp) and employs a paired-end 150 (PE150) sequencing approach, third-generation whole-genome sequencing typically deals with fragments larger than 10 kilobases (Kb), with the ability to extend up to several megabases (Mb). This characteristic confers particular advantages in the detection of large segment structural variations and complex region variations. Furthermore, third-generation whole-genome sequencing obviates the need for PCR amplification, thereby enabling simultaneous extraction of methylation information.
4. Why use long-read sequencing to detect structural variations (SV)?
Structural variants (SV), comprised of deletions, insertions, duplications, and inversions, constitute the majority of variant base pairs in an individual human genome. Many studies have demonstrated a direct or indirect relationship between SVs implicated in human health and their associated phenotypes. Consequently, identifying genetic variants and understanding their functional implications rank among the most critical issues in human genetic research. Given the tendency of SVs to arise within repeat regions and the possible complexity of intra-SV structure, discovering these variants and genotyping can pose significant challenges. Therefore, the need for long-read sequencing technologies to initiate new investigations into SV structures, and to carry out analyses on human population SV detection, is paramount for understanding diseases related to SVs.
5. How is Data Quality Control Conducted?
Data quality control involves several steps to ensure the high quality of sequencing data:
- Evaluation of Read Length Distribution: Inspecting the length distribution of generated reads to ensure it meets expectations.
- Assessment of Error Rate: Evaluating sequencing error rates using internal control sequences or reference genomes.
- Coverage Evaluation: Examining the depth and uniformity of genome coverage to ensure sufficient data coverage.
6. How to Select the Appropriate Sequencing Depth?
The choice of sequencing depth depends on the specific requirements and goals of the study:
- Whole Genome Assembly: Typically requires higher coverage depth to ensure assembly integrity and accuracy.
- Variant Detection: Moderate coverage depth may be sufficient for detecting SNPs and InDels, but detecting CNVs and SVs may require higher coverage.
- Epigenetic Modification Analysis: Coverage depth should be sufficiently high to ensure reliable detection of modification signals.
Human Whole Genome PacBio SMRT Seq Case Studies
Detecting a long insertion variant in SAMD12 by SMRT sequencing: implications of long-read whole-genome sequencing for repeat expansion diseases
Journal: Journal of human genetics
Impact factor: 5.881
Published: 17 December 2018
Background
Short-read next-generation sequencing (NGS) is widely used in medical research and genetic testing to detect pathogenic single-nucleotide variants and small insertions and deletions (indels). However, this technology may miss structural variations (SVs) spanning hundreds to tens of thousands of base pairs. Long-read sequencing technology offers promise in reliably detecting novel SVs. BAFME, an autosomal-dominant neurological disease characterized by tremulous myoclonus and infrequent seizures, is associated with repeat expansions in the SAMD12 gene. PacBio SMRT sequencing, capable of reading >10-kb DNA, has the potential to fully cover the SAMD12 repeat expansion. Long-read whole-genome sequencing using PacBio may be useful for detecting known and novel pathogenic SVs, even with low coverage.
Methods
- Five family members with BAFME
- Peripheral blood leukocytes
- Genomic DNA extraction
- SMRTbell library preparation
- SMRT sequencing
- Sequel Sequencing Kit 2.0.
- SMRT analysis module
- Southern blot analysis
- Dot plot analysis
Results
In a four-generation Japanese family affected by BAFME, a heterozygous structural variant (SV) at the SAMD12 intronic repeat region was identified in the affected individual, consistent with previous studies. The repeat expansion size was similar between individuals after paternal germline passage. PacBio SMRT sequencing was able to fully cover the 4 kb repeat length observed. Long-read whole-genome sequencing (WGS) using the Pacbio Sequel system revealed numerous insertions and deletions, including six SVs specific to the affected individual in the BAFME1-linked region. One notable insertion of 4661 bp was identified between repetitive sequences AluSq2 and (TAAAA)n. Analysis showed that this insertion comprised a novel sequence rather than a tandem duplication, with a high proportion being low-complexity sequences.
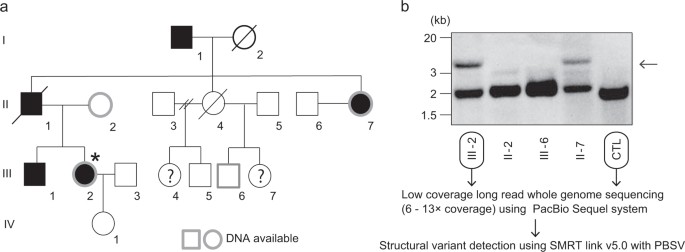 Fig 1. Pedigree of a family with pathogenic structural variation of SAMD12.
Fig 1. Pedigree of a family with pathogenic structural variation of SAMD12.
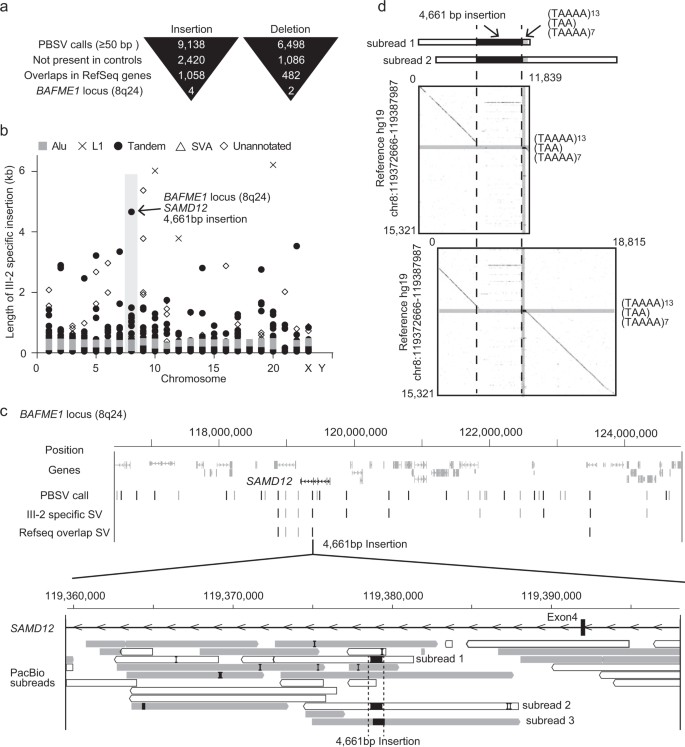 Fig 2. Evaluation of long-read WGS.
Fig 2. Evaluation of long-read WGS.
Conclusion
Long-read sequencing now reads DNA over 10 kb, enabling the detection of large structural variations. The authors applied PacBio SMRT sequencing to a family with benign adult familial myoclonus epilepsy, identifying six SVs in a 7.16-Mb BAFME1 region and confirming a 4.6-kb SAMD12 repeat insertion as causal. Long-read WGS holds promise for comprehensive SV analysis and uncovering new disease-causing variants in undiagnosed conditions.
Reference:
- Mizuguchi T, Toyota T, Adachi H, et al. Detecting a long insertion variant in SAMD12 by SMRT sequencing: implications of long-read whole-genome sequencing for repeat expansion diseases. Journal of human genetics. 2019, 64(3):191-7.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Bacterial communities of Cassiopea in the Florida Keys share major bacterial taxa with coral microbiomes
Journal: bioRxiv
Year: 2024
Production of a Bacteriocin Like Protein PEG 446 from Clostridium tyrobutyricum NRRL B-67062
Journal: Probiotics and Antimicrobial Proteins
Year: 2024
Untangling the Role of Pathobionts from Bacteroides Species in Inflammatory Bowel Diseases
Journal: bioRxiv
Year: 2023
A chromosome-level genome resource for studying virulence mechanisms and evolution of the coffee rust pathogen Hemileia vastatrix
Journal: bioRxiv
Year: 2022
Streptomyces buecherae sp. nov., an actinomycete isolated from multiple bat species
Journal: Antonie Van Leeuwenhoek
Year: 2020
See more articles published by our clients.


