
 Sample Submission Guidelines
Sample Submission Guidelines
![]()
What Is Metatranscriptomic Sequencing
Metatranscriptomic sequencing provides a real-time snapshot of gene activity within microbial communities. Rather than analysing what genes are present (as in metagenomics), this method focuses on what genes are actively expressed under specific conditions—revealing microbial behaviour, regulation, and metabolic output. Whether you're studying the gut microbiome or an industrial fermentation system, metatranscriptomics answers the question:
“What are the microbes doing right now?”
How it works:
- Extract total RNA from the sample (e.g. stool, soil, tissue)
- Remove ribosomal RNA (rRNA) to enrich for messenger RNA (mRNA)
- Convert mRNA into complementary DNA (cDNA)
- Construct sequencing libraries and perform high-throughput sequencing
- Analyse gene expression patterns to reveal active microbial functions
 Metatranscriptomic Sequencing Workflow.
Metatranscriptomic Sequencing Workflow.
Why Choose Metatranscriptomic Sequencing
This next-generation technique offers deep functional insights that DNA-based methods cannot provide. Instead of simply identifying who’s there, it reveals what they’re doing.
- Direct Insight into Microbial Function
Quantifies gene expression across all microbial domains—bacteria, archaea, fungi, and viruses. - Culture-Free, Comprehensive Detection
By bypassing the need for culturing, the method captures real-world community dynamics across all microbes, including non-culturable species. - Dynamic, Time-Resolved Analysis
Compare gene expression across time points or treatment conditions to pinpoint functional changes or identify potential biomarkers. - Supports Mechanistic Discovery
Combined with pathway databases like KEGG, it helps reconstruct metabolic routes and regulatory networks. - Compatible with Multi-Omics
Integrates seamlessly with metagenomics, host transcriptomics, and metabolomics for deeper biological interpretation.

How Does It Compare to Other Microbiome Tools?
| Technique | What It Detects | Reflects Functional Activity? | Resolution | Best For |
|---|---|---|---|---|
| 16S/ITS Amplicon | Marker genes from bacteria/fungi | ❌ No | Medium (Genus/Species) | Rapid screening, taxonomic profiling |
| Metagenomics | All microbial DNA (taxonomy + potential functions) | ❌ No (functional potential only) | High (Strain-level) | Identifying species and potential metabolic capabilities |
| Metatranscriptomics | Actively expressed microbial RNA | ✅ Yes | High (Gene + Strain level) | Expression profiling, mechanism studies, biomarker discovery |
| Host Transcriptomics | Host RNA expression | ✅ Yes | High | Host-microbe interaction studies |
End-to-End Metatranscriptomic Sequencing Service Workflow
Streamlined service from sample to results—maximizing quality and efficiency.

Define goals
Confirm workflow

Register samples
RNA QC
(Optional) RNA extraction

Remove rRNA (>90%)
Build dual-indexed libraries
Perform library QC

Illumina / MGI short reads
PacBio long reads
Customizable depth

Data QC
Transcriptome analysis
Functional annotation
Report generation & delivery
Metatranscriptomic Sequencing Strategy Overview
Supported Sample Types:
- Stool, tissue, saliva, swabs, and more
Library Construction Highlights:
- >90% rRNA depletion
- Unique dual indexing
- Strict quality validation
Sequencing Platforms:
- Illumina NovaSeq X: 150 bp PE – broad expression profiling
- MGI DNBSEQ-G400: 100/150 bp PE – cost-effective transcriptomics
- PacBio Sequel IIe: 15–25 kb HiFi – isoform-level resolution
Recommended Depth:
- Standard: 5–10 Gb/sample
- Optional: Higher depth for low-abundance transcripts
Data Quality Metrics:
- >80% bases at Q30+
- Error rate <0.1%
- Accurate, reliable results
Metatranscriptomic Bioinformatics Analysis
Complete data analysis from raw reads to publication-ready visuals, supporting your research and grant needs.
Microbial Expression Profiling
- Profile active gene expression across bacteria, fungi, viruses, and archaea
- Visualize species abundance and diversity metrics
Functional Annotation & Pathway Analysis
- Annotate key genes using UniRef and UniProt
- Reconstruct metabolic pathways with KEGG and MetaCyc
- Perform differential pathway expression analysis
Antibiotic Resistance & Virulence Detection
- Detect antibiotic resistance genes (AMR) and virulence factors
- Quantify expression and analyze pathway enrichment
Publication-Ready Visuals & Reports
- High-quality PCA, heatmaps, volcano plots, and more
- Statistical analysis including LEfSe for key functional differences
- Expression data normalized and provided in easy-to-use tables

Sample Requirements for Metatranscriptomic Sequencing
To ensure optimal sequencing performance, samples must meet baseline quantity and purity standards. Custom consultation is available for specialised sample types.
| Sample Type | Minimum Requirements |
|---|---|
| Total RNA | ≥ 4 μg (≥ 3 μg minimum), ≥ 50 ng/μL |
| Cultured Cells | ≥ 5 × 10⁶ cells |
| Environmental Samples | ≥ 1.5 grams |
📩 Not sure if your sample is suitable? Contact us for personalised pre-treatment guidance.
Is Metatranscriptomics Right for My Research
Metatranscriptomic sequencing reveals what your microbiome is actually doing, not just who’s there. If your study involves microbial activity, gene expression dynamics, or functional pathway shifts, this technique may be a perfect fit.
Ideal Use Cases:
- Health & Disease Microbiome Studies
Track how gut, skin, or respiratory microbiomes behave under healthy or diseased states. - Drug, Diet, or Environmental Interventions
Quantify how treatments impact microbial gene expression and metabolic pathways. - Environmental & Agricultural Microbial Ecology
Assess the active functions of microbes in soil, water, or host-associated systems. - Probiotic & Microbiome Therapeutics Development
Identify beneficial strains and validate their functional mechanisms in action. - Unculturable Microbe Discovery
Detect active expression from hard-to-culture organisms missed by traditional methods.
Common Research Questions We Help Answer:
- How does microbial gene activity shift in response to a treatment or condition?
- Which genes or pathways are activated after a dietary or pharmaceutical intervention?
- Can I uncover new functions in microbes that can’t be cultured in the lab?
- How can I functionally characterise strains for probiotic or biomarker development?
Combine Metatranscriptomics with Other Omics for Deeper Insights
| Paired Technique | Combined Advantage | Example Application |
|---|---|---|
| Metagenomics + Metatranscriptomics | Identify both potential and actual gene activity | Differentiate silent vs. active strains in microbial communities |
| Host Transcriptomics + Metatranscriptomics | Decode host–microbe interaction networks | Investigate inflammation/infection models |
| Metabolomics + Metatranscriptomics | Link gene expression to real metabolic output | Explore drug/diet impact on microbial metabolism |
| 16S/ITS + Metatranscriptomics | Screen large cohorts, then zoom into active samples | Efficient sample triage before deep functional profiling |
Why Choose CD Genomics for Metatranscriptomic Sequencing?
When it comes to capturing microbial gene expression with precision and depth, CD Genomics offers more than just sequencing—we deliver actionable insights backed by years of experience and full-service support.
- Extensive Multi-Omics Expertise
Trusted by top-tier research institutes and biotech companies, we bring a solid track record in transcriptomics, genomics, and microbiome profiling. - Flexible Platform Options
Choose from Illumina, MGI, or PacBio platforms to match your sample type, budget, and resolution needs. - Customised Bioinformatics Analysis
Gain deeper insights through advanced analytics including functional annotation, pathway enrichment, and differential expression mapping. - Stringent Quality Control at Every Step
From sample handling to final reporting, our end-to-end traceability system ensures reliable, reproducible results. - Expert Support From Start to Finish
Our technical team offers real-time guidance and troubleshooting, helping you accelerate timelines and overcome project challenges.

Demo Results
Partial results are shown below:
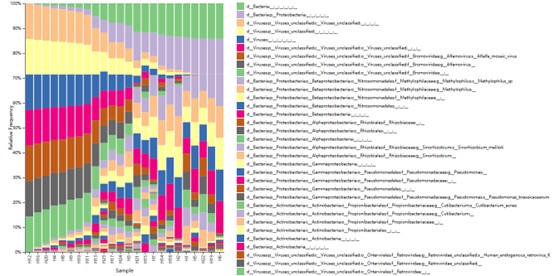
The taxonomy distribution of all sample in Phylum classification level.
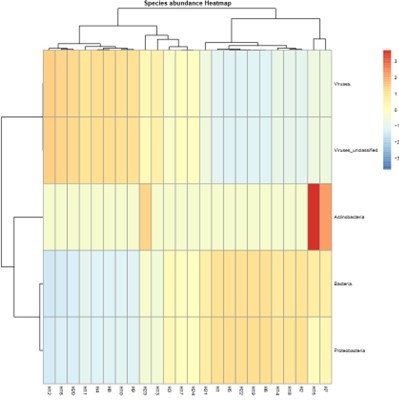
Species abundance Heatmap.
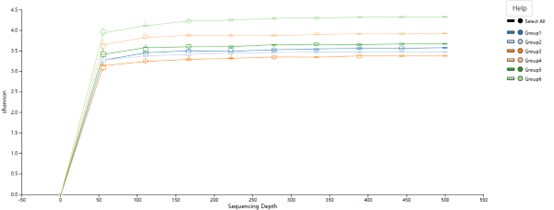
Rarefaction curve of the sequenced reads for all samples.
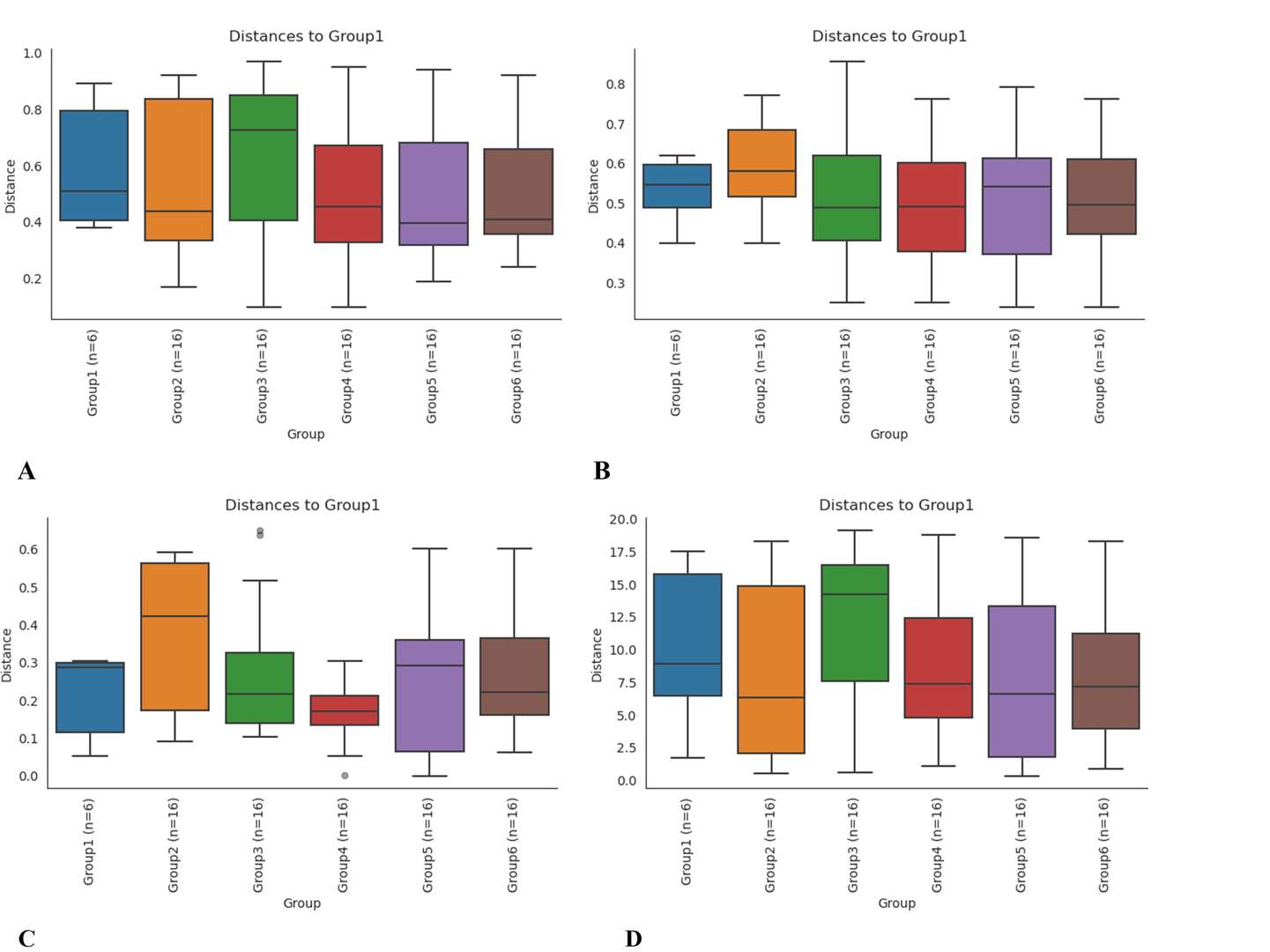
Boxplot analysis based on bray Curtis (A), binary jaccard (B), unweighted unifrac (C), and weighted unifrac (D).
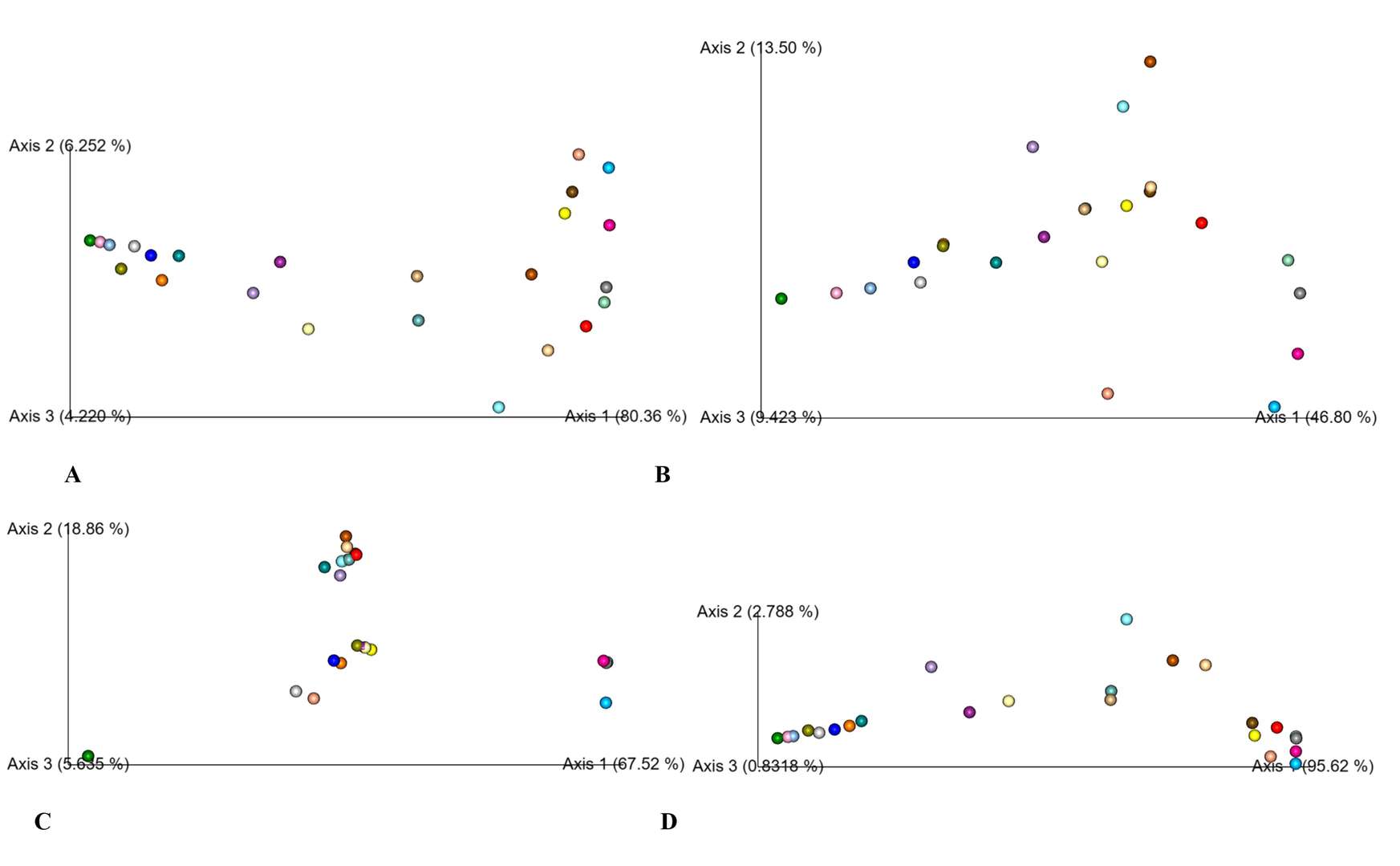
PCoA analysis based on bray Curtis (A), binary jaccard (B), unweighted unifrac (C), and weighted unifrac (D).
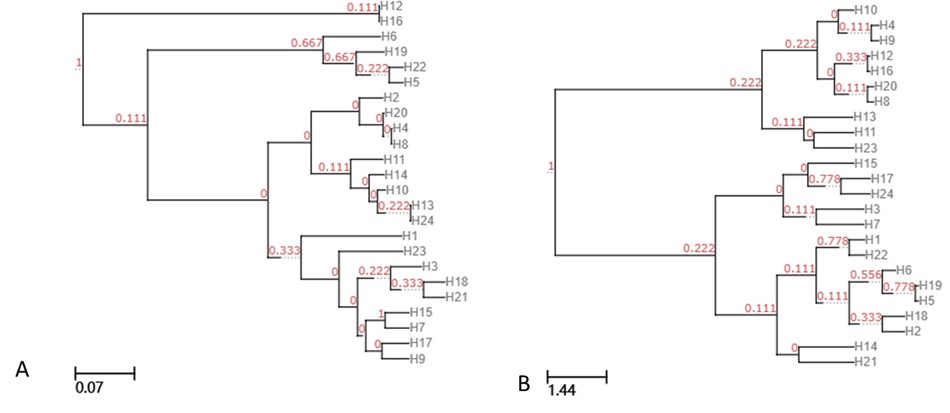
UPGMA clustering tree based on unweighted unifrac (A), and weighted unifrac (B).
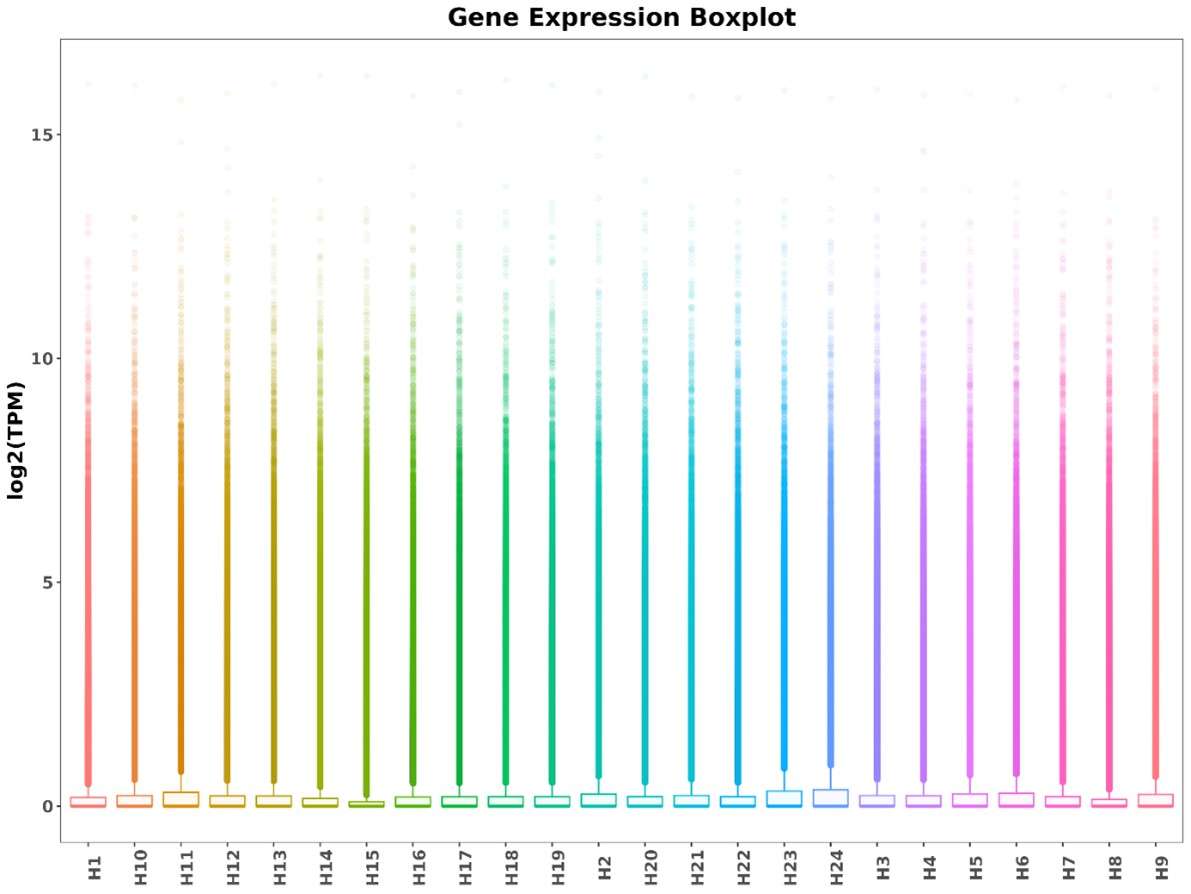
Boxplot of TPM for each sample.
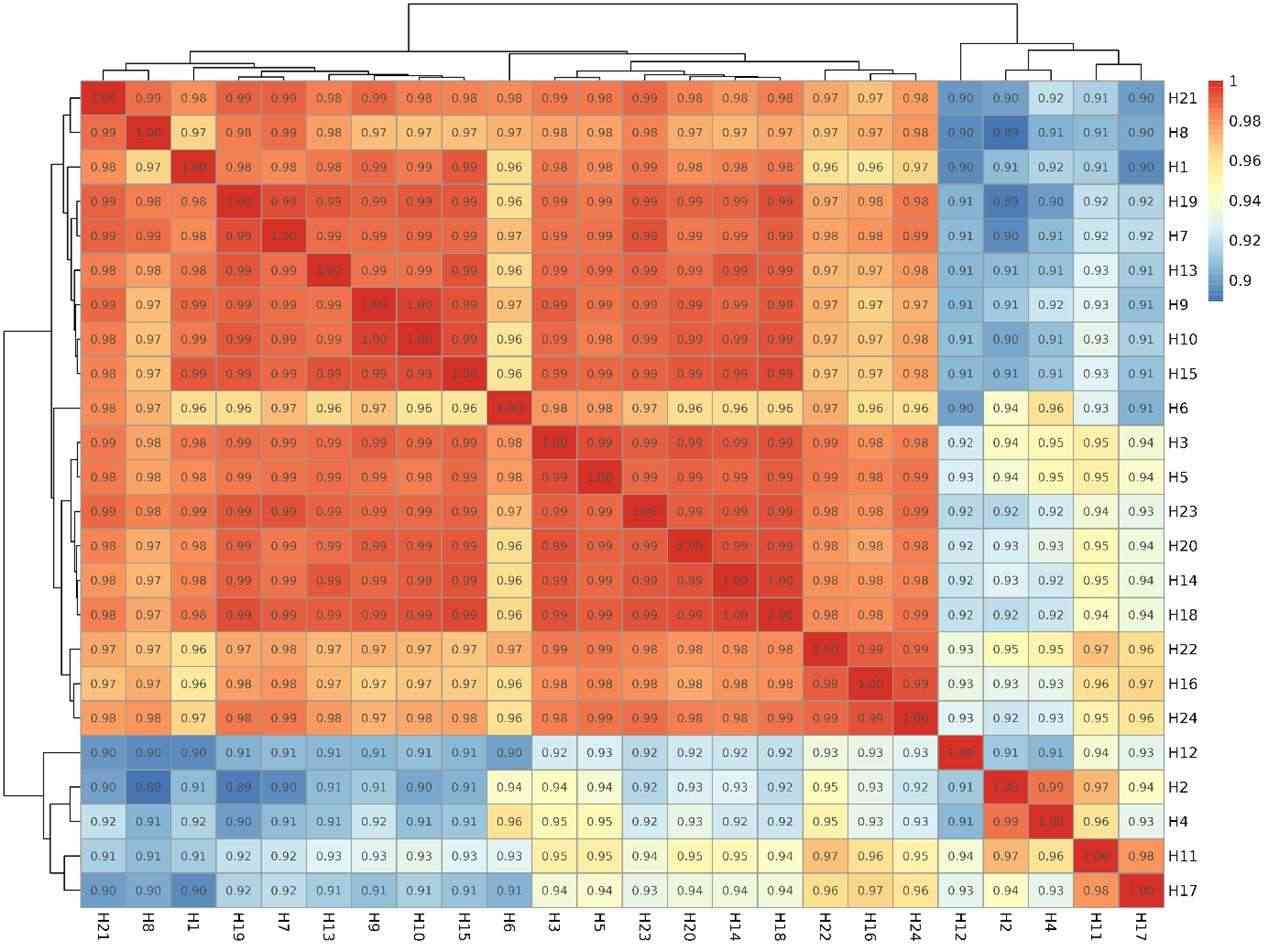
Correlation graph of gene number.
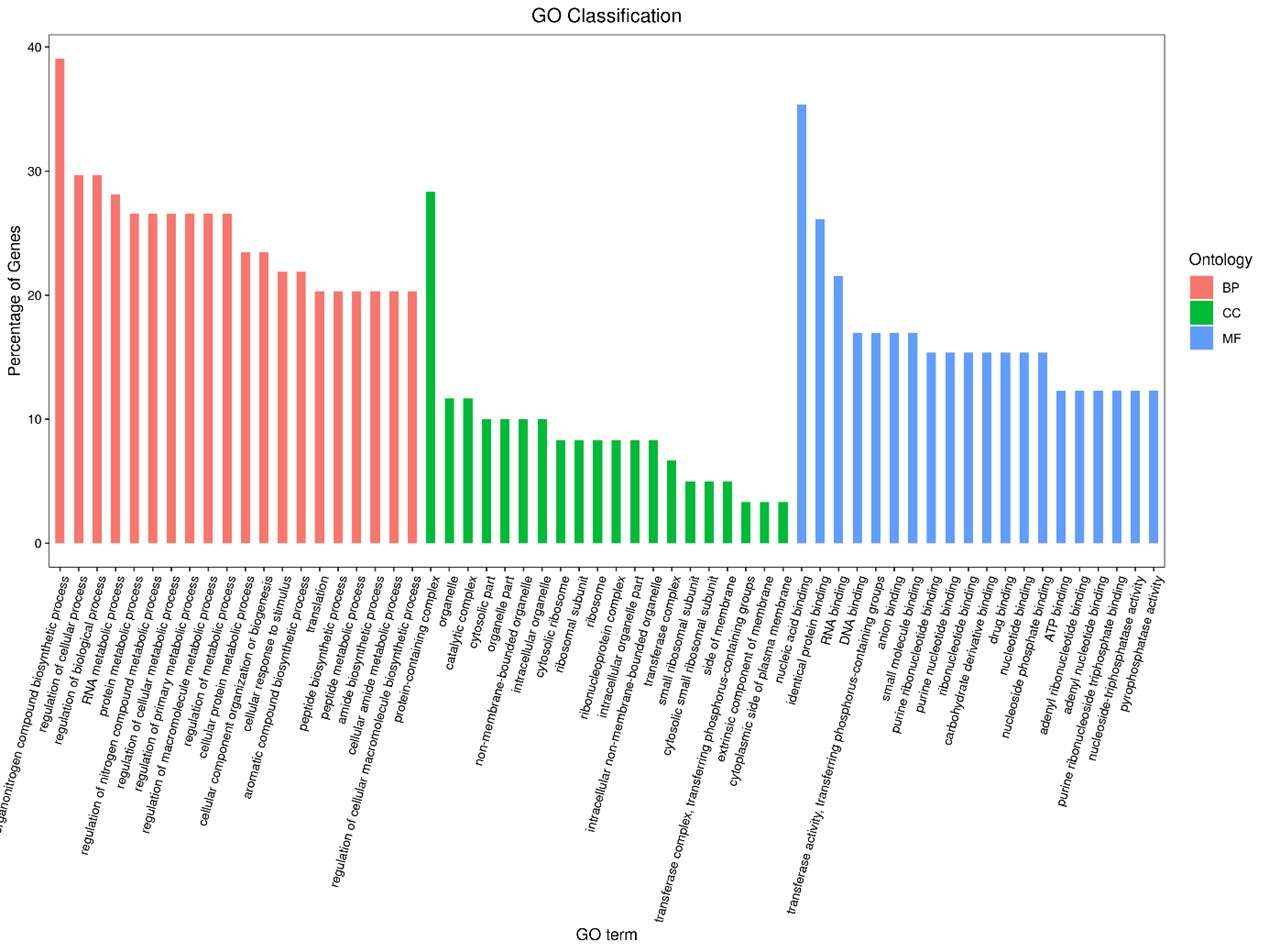
Statistics results of GO annotation for CLC_vs_SLC.
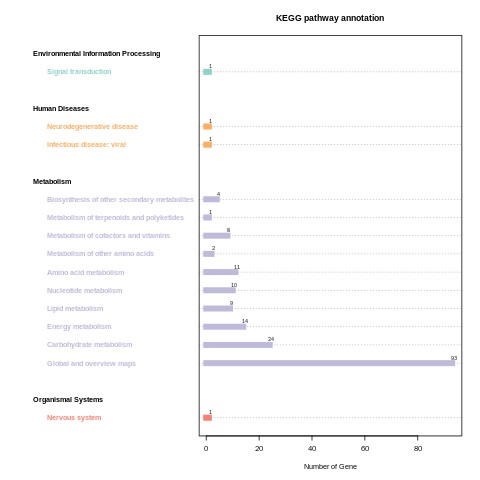
CLC_vs_SLC KEGG_classification.

Statistical of specific function database common and unique annotation.

CAZy function classification.
Metatranscriptomic Seq FAQs
1. What are the noteworthy issues of RNA samples?
The contamination should be rigorously excluded when sampling. In detail, sampling-related instruments and consumables should be sterilized and RNase-free. The freshly obtained samples should be immediately frozen by putting into liquid nitrogen, or directly submitting original environmental or clinical samples to us. The recommended total RNA amount for submission is 6 µg or more with a concentration of greater than 50 ng/µl.
2. What kind of QC methods do you adopt for the customer's samples?
We will perform QC on your total RNA samples prior to sequencing them. We use the Agilent Bioanalyzer to determine the RNA Integrity Number (RIN). If the RIN is lower than 8, the samples will not pass QC. The library QC will also be performed using the Agilent Bioanalyzer to determine library size and purity. Also, prior to loading the libraries on the sequencer, we perform qPCR quantification. The cost for this is included in the sequencing service. The raw data will pass our Q30 filter, which means more than 80% of bases with a greater than Q30 quality score.
3. What are the advantages of metatranscriptomics?
Metatranscriptomics is the genomic analysis of complete microbial transcriptomes, providing a particularly rich source of data on the global diversity of RNA viruses and their evolutionary history. Metatranscriptomics has several advantages over traditional methods such as cell culture, consensus PCR, and metagenomics approaches based on viral particle purification.
Metatranscriptomics has proven successful in characterizing the RNA viromes of diverse invertebrates. Specifically: (i) it uncovers the entire RNA virome, with sufficient coverage to assembly complete viral genomes, including those from co-infecting parasites; (ii) it offers a reliable quantification and assessment of both viral and host RNAs; (iii) it is comparatively simple, requiring minimal sample processing; and (iv) it provides more information than the genome sequence alone, allowing a characterization of viral diversity and ecology.
4. I’m unsure if my samples are suitable for metatranscriptomics. Can you assess them first?
Absolutely. We offer free feasibility assessments based on your study objectives and sample type. Before sequencing begins, we’ll recommend the best platform, depth, and analytical strategy tailored to your goals.
5. Can I integrate metatranscriptomics with metagenomics or other omics datasets?
Yes, we specialise in multi-omics integration. Whether you're combining with metagenomics, metabolomics, or host transcriptomics, our team can build a unified analytical workflow to uncover functional and taxonomic insights across datasets.
References
- Shi M, Neville P, Nicholson J, et al. High-Resolution Metatranscriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia. Journal of Virology, 2017, 91(17): e00680-17.
- Shi M, Zhang Y Z, Holmes E C. Meta-transcriptomics and The Evolutionary Biology of RNA Viruses. Virus research, https://doi.org/10.1016/j.virusres.2017.10.01
Metatranscriptomic Seq Case Studies
Customer Publication Highlight
Hydrogen-Oxidizing Bacteria Are Abundant in Desert Soils and Strongly Stimulated by Hydration
Journal: mSystems
Published: 2020
DOI: 10.1128/mSystems.01131-20
Background
Desert soils sustain diverse bacterial communities despite extreme aridity. While photosynthesis was traditionally considered the primary energy source, recent evidence suggests atmospheric trace gases (e.g., H₂) may support microbial survival. This study investigated the role of hydrogen-oxidizing bacteria across four global deserts (Australian, Namib, Gobi, Mojave), revealing unprecedented H₂ oxidation rates stimulated by hydration and its coexistence with photosynthesis.
Project Objectives
- Metabolic Profiling: Quantify distribution/activity of hydrogenases and photosystems.
- Hydration Response: Assess microbial activity shifts during wet-dry cycles.
- Cross-Desert Validation: Compare H₂ oxidation in polar vs. nonpolar deserts.
CD Genomics’ Services
As a genomic analytics partner, CD Genomics enabled:
- Metagenomic & Metatranscriptomic Sequencing
- Platform: Illumina NovaSeq (shotgun metagenomics) + Oxford Nanopore (long-read MAG assembly).
- Coverage: 563M read pairs for Australian desert soil; multi-omics for hydration time-series.
- Library Prep: Dual-indexed libraries from 0–10 cm depth soil; rRNA depletion for transcriptomes.
- Bioinformatics Analysis
- Assembly & Binning: MetaSPAdes v3.15; MaxBin2 for 39 metagenome-assembled genomes (MAGs).
- Functional Annotation:
- HydDB for hydrogenase classification (groups 1h, 1l, 2a).
- KEGG/MEROPS for respiration, photosynthesis, and carbon fixation pathways.
- Variant Analysis: SNP calling in hydrogenase genes across continents.
- Activity Validation
- Gas chromatography (GC) for H₂ consumption rates (Fig. 3).
- Isotopic labeling (¹³C-CO₂) to quantify carbon fixation.
Key Findings
- Ubiquitous Hydrogenase Genes
- Hydrogenase sequences dominated metagenomes (45% of community), prevalent in Actinobacteriota (39%), Proteobacteria (17%), and Cyanobacteria (3.2%).
- First report of group 2a [NiFe]-hydrogenases in desert cyanobacteria (Nostoc, Tolyopthrix).
- Hydration-Driven Metabolic Surge
- H₂ oxidation rates increased 950-fold post-hydration (Fig. 3c).
- Photosynthesis and dark carbon fixation rose 3-fold and 1.7-fold, respectively.
- Global Desert Conservation
- Hydrogenase genes confirmed in all four deserts. H₂ oxidation was simultaneously activated with photosynthesis upon wetting, debunking prior "alternating energy mode" hypotheses.
Figures Referenced
 FIG 3 H2 oxidation by Australian desert soil microcosm samples.
FIG 3 H2 oxidation by Australian desert soil microcosm samples.
 Fig. 2: Heatmaps showing hydrogenases (groups 1h/1l/2a) as most abundant respiratory genes. Expression persisted even after hydration (144 TPM in dry soils; stable in wet soils).
Fig. 2: Heatmaps showing hydrogenases (groups 1h/1l/2a) as most abundant respiratory genes. Expression persisted even after hydration (144 TPM in dry soils; stable in wet soils).
Implications
- Ecological Modeling: H₂ oxidation is a major energy source for desert microbiomes, revising carbon/energy flux models in arid ecosystems.
- Climate Resilience: Hydration-responsive bacteria could engineer drought-tolerant soil communities for desert restoration.
- Biogeochemical Impact: Global H₂ consumption by deserts may influence atmospheric gas budgets.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Transferrable protection by gut microbes against STING-associated lung disease
Journal: Cell Reports
Year: 2021
Microbial adaptation and response to high ammonia concentrations and precipitates during anaerobic digestion under psychrophilic and mesophilic conditions
Journal: Water Research
Year: 2021
Algal-bacterial synergy in treatment of winery wastewater
Journal: NPJ Clean Water
Year: 2018
Black soldier fly bioconversion to cultivated meat media components using blue catfish gut microbiome
Journal: Bioresource Technology Reports
Year: 2024
Indole-3-Propionic Acid, a Gut Microbiota Metabolite, Protects Against the Development of Postoperative Delirium
Journal: Annals of Surgery
Year: 2023
Elucidating the effects of organic vs. conventional cropping practice and rhizobia inoculation on rhizosphere microbial diversity and yield of peanut
Journal: Environmental Microbiome
Year: 2023
See more articles published by our clients.


