
 Sample Submission Guidelines
Sample Submission Guidelines
CircRNA Sequencing
To support the recent emerging of circular RNA (circRNA) research, CD Genomics is providing the circRNA sequencing service that utilizes the latest Illumina platforms for a fast, economical, and accurate characterization.
The Introduction of CircRNA Sequencing
CircRNA is a novel member of the non-coding RNAs (ncRNAs), generated by non-sequential backsplicing of exons, introns or both. They are characterized by covalently closed loop feature thus lacking 5' end caps and 3' poly-A tails. CircRNAs are highly stable forms of ncRNAs due to their nuclease resistance properties. When first identified in the 1970s, circRNAs were regarded as viral genomes or byproducts of mis-splicing events. But the recent researches have revealed that circRNAs are evolutionarily conserved across plants, animals, and human beings, and that circRNAs have important biological functions.
CircRNAs may act as miRNA sponges and fulfill a regulatory function in gene expression. Remarkably, a wide range of circRNAs are abnormally expressed in specific disease contexts, which suggests their association with the occurrence and development of human diseases. CircRNAs also perform multiple functions in cellular processes including templates for translation, regulation of alternative splicing and gene expression, scaffolds for the assembly of protein complexes, and modulator of rRNA and tRNA biogenesis. Additionally, they can be used to boost immune activation for antiviral therapeutic purposes due to their intervention in immune regulation and viral infection.
Comprehensive detection of circRNAs from high-throughput transcriptome data is an initial and crucial step to study their biogenesis and function. The high-throughput sequencing of rRNA/linear RNA-depleted RNA in combination with computational tools has led to the identification of thousands of new circRNAs and analysis of their linear host transcripts in a quantitative manner in diverse organisms. Unlike miRNAs and other small RNAs, circRNAs are not easily separated from other RNA species by size or electrophoretic mobility. Consequently, they are generally retained in rRNA-depleted libraries and enriched in libraries treated with RNase R that only digest linear RNA.
Advantages of CircRNA Sequencing
- Identifies known and novel circRNAs
- Allows profiling of circRNAs across a wide dynamic ranges
- Explores novel biomarkers and circRNAs regulatory networks
Applications of CircRNA Sequencing
CircRNA sequencing can be used for but not limited to the following research:
- Investigating the pathogenic mechanisms of diseases such as cancer;
- Exploring gene expression regulatory changes during growth and development;
- Researching other aspects related to gene transcription and expression regulation.
CircRNA Sequencing Workflow
The general workflow for circRNA sequencing is outlined below. To construct circRNA sequencing library, the first step is to deplete rRNA, followed by linear RNA digestion and strand-specific library preparation. Our highly experienced expert team executes quality management, following every procedure to ensure confident and unbiased results.

Service Specification
Sample Requirements
|
|
 Click |
Sequencing Strategies
|
 |
Data Analysis We provide multiple customized bioinformatics analyses:
Note: Recommended data outputs and analysis contents displayed are for reference only. For detailed information, please contact us with your customized requests. |
Analysis Pipeline
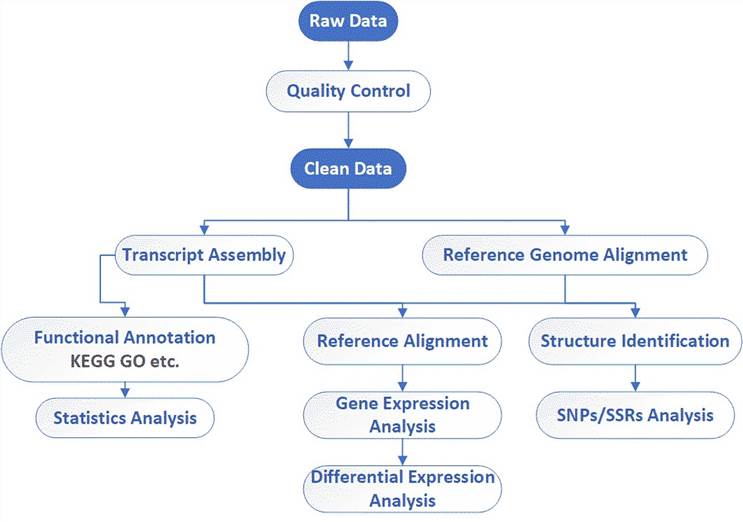
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in CircRNA Sequencing for your writing (customization)
Supported by our experienced scientists and advanced technology, CD genomics can help you to acquire the circRNA sequence information with single-base resolution at one time through the high-throughput sequencing by strict quality control and advanced bioinformatics analyses. If you have additional requirements or questions, please feel free to contact us.
Reference:
- Wang M, Yu F, Wu W, et al. Circular RNAs: A novel type of non-coding RNA and their potential implications in antiviral immunity. International journal of biological sciences, 2017, 13(12): 1497.
Demo Results
Partial results are shown below:

Sequencing quality distribution

A/T/G/C Distribution

IGV Browser Interface

Correlation Analysis Between Samples

PCA Score Plot

Venn Diagram

Volcano Plot

Statistics Results of GO Annotation

KEGG Classification
CircRNA Seq FAQs
1. How is circRNA generated?
CircRNAs are generally generated through the back-splicing of exons in a non-sequential order. Circularization of RNA can be generated by repetitive sequences to facilitate the back-splicing of non-sequential exons.
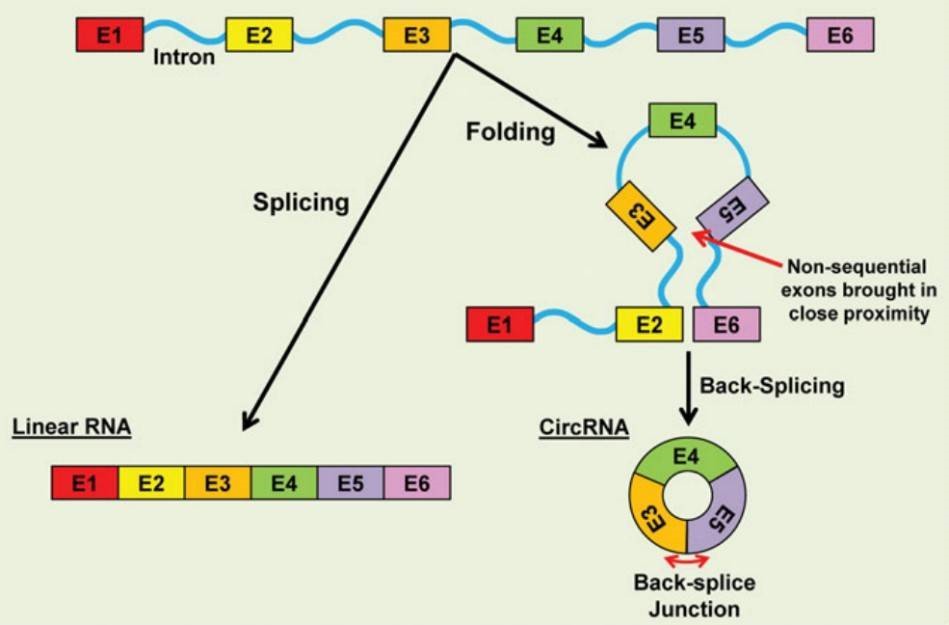 Figure 1. CircRNA biogenesis (Fischer & Leung 2016).
Figure 1. CircRNA biogenesis (Fischer & Leung 2016).
2. Why using high-throughput technology to analyze circRNAs over microarrays?
High-throughput sequencing is a time and cost saving method with many advantages over DNA microarrays.
i. Detection of novel transcripts. Microarrays depend on the hybridization with species or transcript-specific probes that are restricted to known circRNAs. However, RNA sequencing can detect novel transcripts, gene fusions, single nucleotide variants, small insertions and deletions, and other previously unknown changes.
ii. Broader dynamic range. Microarrays are limited by background at the low end and signal saturation at the high end, while RNA sequencing can quantify discrete, digital sequencing read counts, offering a broader dynamic range.
iii. Increased sensitivity and specificity. Compared to microarrays, RNA sequencing offers increased sensitivity and specificity, enabling enhanced detection of genes, transcripts, and differential expression.
iv. Detection of low-abundance and rare transcripts. Sequencing coverage depth can easily be increased for detection of rare transcripts, single transcripts per cell, or weakly expressed genes.
3. Are there any tips for constructing circRNA expression vector of circRNA?
Yes. Sequences for circularization and sequences that activate the circularization are required to construct circRNA expression vector carrying circRNAs.
Reference:
- Fischer J W, Leung A K L. CircRNAs: a regulator of cellular stress. Critical reviews in biochemistry and molecular biology, 2017, 52(2): 220-233.
CircRNA Seq Case Studies
CircHLA-C plays an important role in lupus nephritis by sponging miR-150
Journal: Molecular Therapy: Nucleic Acid
Impact factor: 6.392
Published: January 12, 2018
Abstract
Circular RNAs participate in the pathogenesis of multiple diseases by acting as miRNAs sponges. The previous studies have reported that miR-150 positively correlated with renal chronicity index in patients with lupus nephritis (LN). The authors further investigated renal circRNA profiling and the interaction between cirRNAs and miR-150 in LN patients. The bioinformatics analysis predicted that miR-150 was regulated by circHLA-C and that circHLA-C probably played an important role in the pathogenesis of LN by sponging miR-150.
Materials & Methods
- Human kidney tissue samples
- Seven LN patients
- RNA extraction
- Library construction
- circRNA sequencing
- HiSeq 4000 sequencing system
- Real-time qPCR
- Hierarchical clustering analysis
- Differential expression analysis
- GO and KEGG analysis
- Statistical analysis
Results
1. Profiling and characteristic of circRNAs in renal biopsies of LN patients
Total 18505 circRNA transcripts were identified including 11411 up-regulated and 7094 down-regulated circRNAs. Hierarchical clustering heatmap showed that the circRNAs expression levels were clustered between LN renal biopsies and normal control kidneys. 171 circRNAs were found to significantly express differentially between LN group and normal control group.
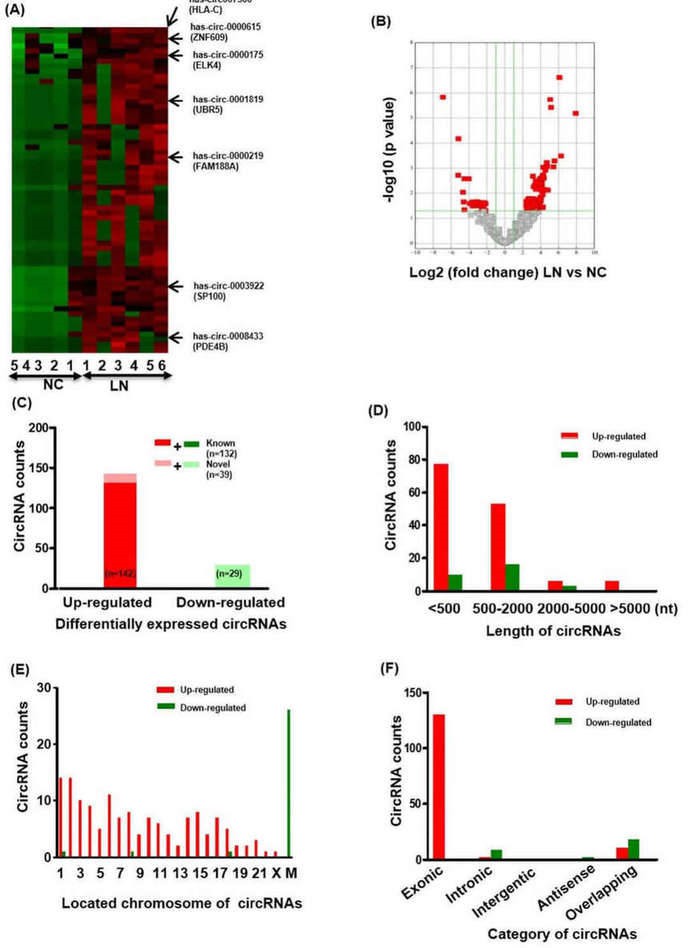 Figure 1. The profiling and characteristics of circRNAs. (A) Clustered heatmap. The red represents upregulated circRNAs and the green represents downregulated circRNAs. (B) Volcano plot. (C-F) The characteristics of differentially expressed circRNAs.
Figure 1. The profiling and characteristics of circRNAs. (A) Clustered heatmap. The red represents upregulated circRNAs and the green represents downregulated circRNAs. (B) Volcano plot. (C-F) The characteristics of differentially expressed circRNAs.
2. The CircRNAs/miRNAs interaction network
The CircRNAs/miRNAs interaction network was predicted by conserved seed-matching sequence. It revealed that all of 40 circRNAs contained their respective miRNAs response elements (MREs). The top five miRNAs regulated by each of the 40 circRNAs were displayed as a network (Figure 2).
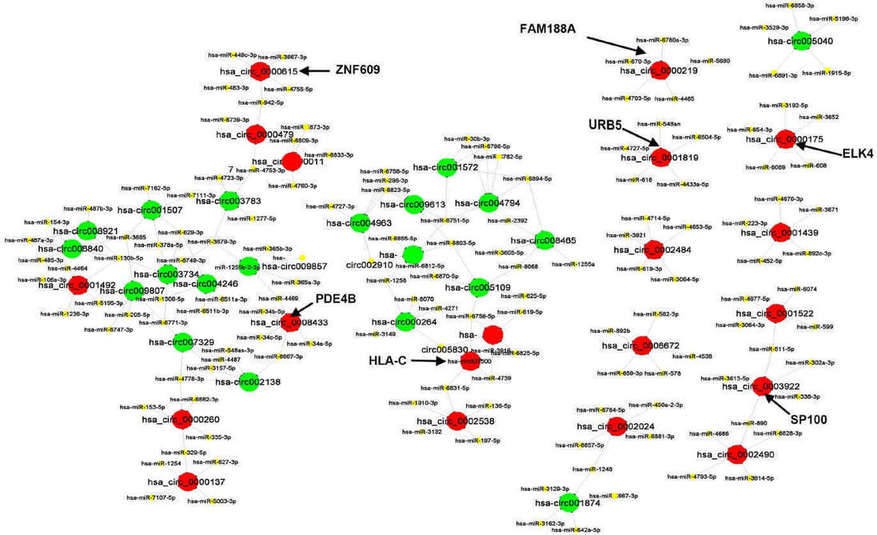 Figure 2. The interaction network between circRNAs and miRNAs.
Figure 2. The interaction network between circRNAs and miRNAs.
3. Prediction of functions and pathways
The functions of differentially expressed circRNAs were predicted by analysis of GO and KEGG. GO enrichment analysis revealed that the functional roles of target host genes were linked to biological processes, cellular components, and molecular functions. KEGG analysis found 23 pathways related to the functions of 142 upregulated circRNAs and both hypoxia inducible factor-1 (HIF-1) signaling pathway and neurotrophin signal pathway were participated in regulating the expression of NF-kB.
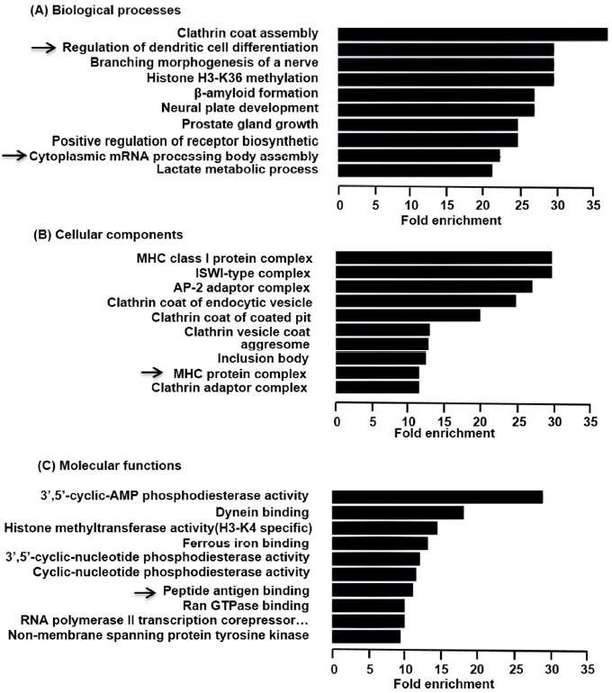 Figure 3. Predicted functions of the overexpressed circRNAs in LP.
Figure 3. Predicted functions of the overexpressed circRNAs in LP.
4. Interaction between circHLA-C and miR-150
CircHLA-C positively correlated with the parameters of LN disease activity. And miR-150 exhibited a tendency of negative correlation with circHLA-C (Figure 4).
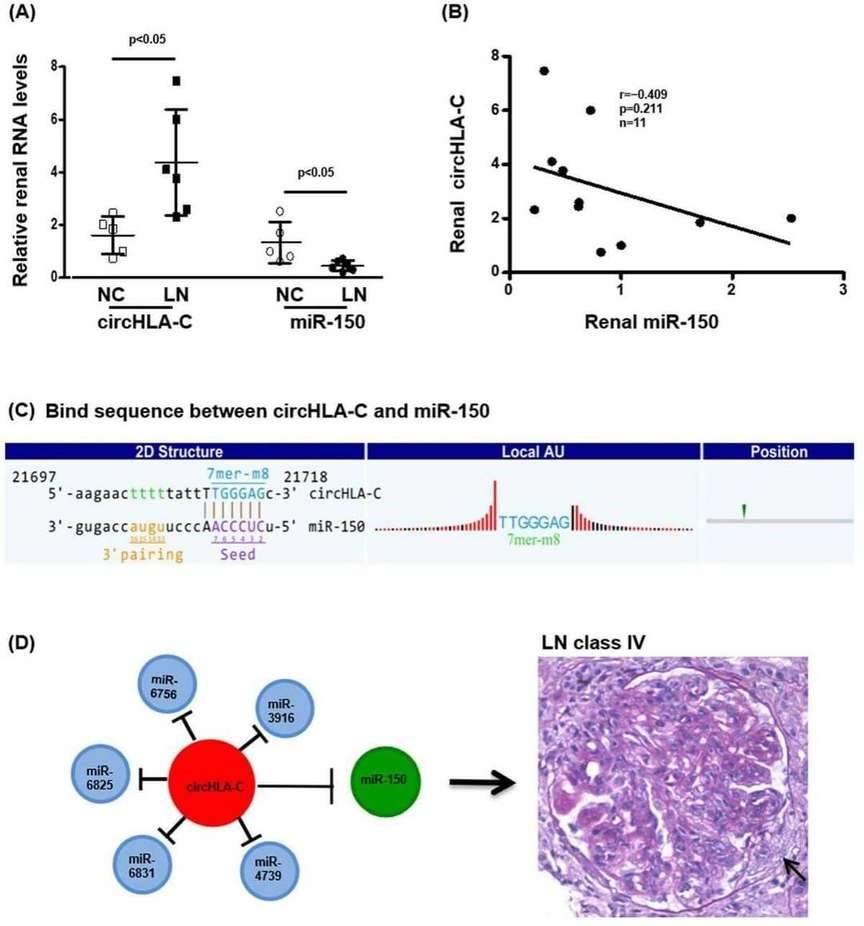 Figure 4. The interaction between renal circHLA-C and miR-150 in LN.
Figure 4. The interaction between renal circHLA-C and miR-150 in LN.
Conclusion
The authors identified 171 differentially expressed circRNAs and validated seven significantly upregulated circRNAs in kidneys of LN class IV patients, with circHLA-C positively correlating with LN disease activity. circHLA-C was significantly increased while miR-150 was decreased, displaying a negative correlation and a perfect binding sequence match. The study also provided the first renal circRNA profiling and interaction network between circRNAs and miRs in LN.
Reference:
- Luan J, Jiao C, Kong W, et al. circHLA-C Plays an Important Role in Lupus Nephritis by Sponging miR-150. Molecular Therapy-Nucleic Acids, 2018, 10: 245-253.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Merkel Cell Polyomavirus Encodes Circular RNAs (circRNAs) Enabling a Dynamic circRNA/microRNA/mRNA Regulatory Network
Journal: Mbio
Year: 2020
Identification of circular RNAs regulating cardiomyocyte proliferation in neonatal pig hearts
Journal: JCI insight
Year: 2024
Circular DNA tumor viruses make circular RNAs
Journal: Proceedings of the National Academy of Sciences
Year: 2018
KMT2A associates with PHF5A-PHF14-HMG20A-RAI1 subcomplex in pancreatic cancer stem cells and epigenetically regulates their characteristics
Journal: Nature communications
Year: 2023
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch
Journal: Science Advances
Year: 2024
Genomic imprinting-like monoallelic paternal expression determines sex of channel catfish
Journal: Science Advances
Year: 2022
See more articles published by our clients.


