
 Sample Submission Guidelines
Sample Submission Guidelines
Genetic Linkage Map
To address the emerging needs of research communities, CD Genomics has developed an affordable, reliable genetic linkage map service based on high-throughput sequencing to get dense markers and give researchers a high-quality genetic linkage map, as well as professional data analysis.
The Introduction of Genetic Linkage Map
Genetic linkage map, also known as genetic map, is a linear graph of the sequence and relative distance of molecular markers on chromosome based on the frequencies of recombination between markers during crossover of homologous chromosomes. The construction of high-density genetic map based on high-throughput sequencing technology has gradually become a revolutionary technology favored by researchers. It can rapidly develop a large number of molecular markers at one time, and obtain ultra-high-density genetic map. It provides accurate and complete QTLs number and locus information that co-segregate with the phenotype.
If you want to learn more about genetic linkage maps, you can read our article "Genetic Linkage Mapping: Definition, Techniques, and Applications".
 Figure 1. A genetic linkage map (Yuhui Zhao, et al. 2020)
Figure 1. A genetic linkage map (Yuhui Zhao, et al. 2020)
What are the Advantages of Genetic Linkage Map
- Understanding Genetic Inheritance
- Gene Identification and Mapping
- Marker-Assisted Selection
- Genetic Diversity Studies
- Facilitating QTL Analysis
- Genomic Research and Development
- Enhancing Crop and Livestock Production
- Support for Functional Genomics
- Improving Genetic Counseling and Disease Prediction
- Advancing Personalized Medicine
- High speed, high density, high quality
- High-quality sequencing data, high-performance computing platform
What are the Application of Genetic Linkage Map
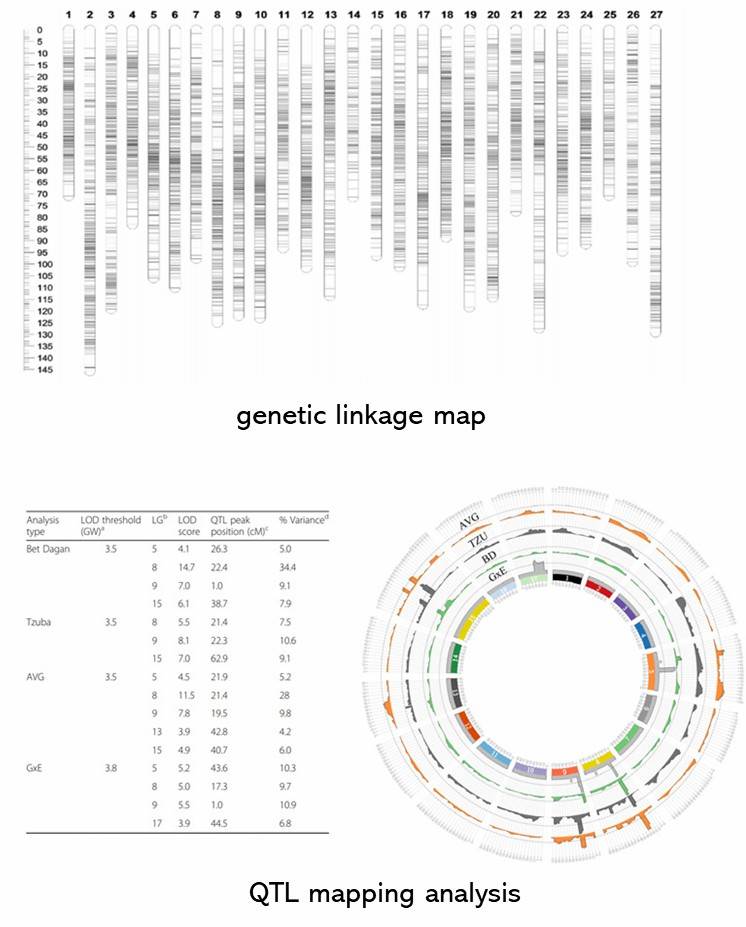
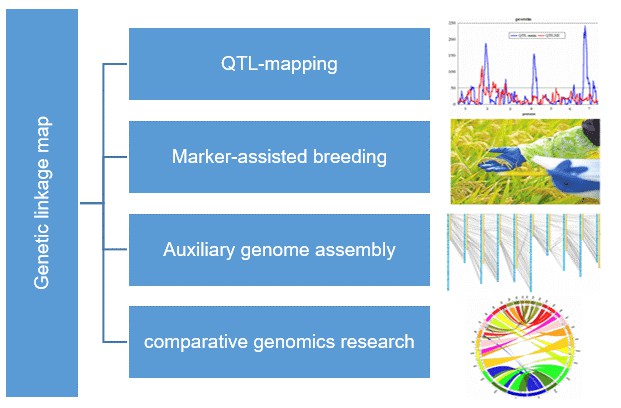 Figure 1. A genetic linkage map (Yuhui Zhao, et al. 2020)
Figure 1. A genetic linkage map (Yuhui Zhao, et al. 2020)
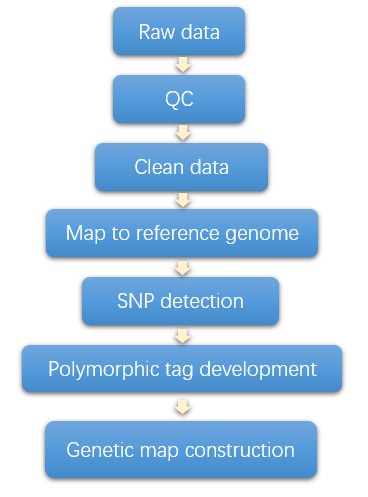
Genetic Linkage Map Workflow

Service Specification
Sample Requirements
|
|
 |
Sequencing
|
 |
Bioinformatics Analysis
We provide customized bioinformatics analysis including:
|
Analysis Pipeline
Deliverables
- Raw data(FASTQ)
- Marker information
- Genetic linkage map evaluation report
- Data analysis report
Reference:
- Zhao YH, et al. High-density genetic linkage-map construction and QTL mapping for important fruit traits. PLoS ONE. 2020,15(2): e0229020.
Demo Results
Genetic Linkage Map FAQs
1. What is a genetic linkage map?
Essentially, a genetic linkage map is a diagrammatic representation that delineates the relative positions of genes or genetic markers on a chromosome, derived from their respective recombination frequencies. This sort of map provides an indispensable visual guide to the order and space between genetic loci. It serves the twofold purpose of shedding light on the complexities of genetic inheritance and informing practicable approaches in fields as diverse as crop breeding, gene mapping, and expansive genetics research.
2. How is a genetic linkage map constructed?
The elaboration of a genetic linkage map generally follows the sequence of steps enumerated below:
- Selection of an Appropriate Population: Frequently chosen populations consist of F2, backcross populations, or recombinant inbred lines.
- Genotyping Implementation: The identification of genetic markers employs technology such as SNP arrays or comprehensive whole-genome resequencing.
- Data Processing and Analytical Evaluation: Through the utilization of both computational tools and statistical methodologies, recombination frequencies between markers are computed, enabling the creation of linkage groups and, subsequently, the construction of the genetic map.
3. How should parental lines be selected?
The selection of parental lines directly influences the difficulty level involved in map construction and the applicability range of the constructed map. It needs to meet the following criteria: (i) Genetic polymorphism between the parental lines. (ii) Consideration for the fertility of the offspring to prevent skewed segregation. (iii) High purity parental line materials should be chosen whenever possible (excluding the F1 generation).
4. What are some software tools for constructing maps?
JoinMap is a software tool that operates on the Widows operating system and is currently the most widely used due to its applicability to virtually all population types. Other tools include R/qtl, Lepmap, Highmap, Onemap, Mstmap, and Carthagen.
5. What is recombination frequency and why is it important?
Indeed, recombination frequency serves as an essential parameter when attempting to delineate the underlying genetics of an organism. It refers to the percentage of progeny in which a specific crossover event between two genes or markers has transpired during the process of meiosis. Manifestly, a higher recombination frequency is indicative of an increased genetic distance separating the genes in question. The importance of this frequency lies in its indispensable role in the construction of genetic linkage maps - these frequencies aid in ascertaining the relative positions of genes within a chromosome. Therefore, the recombination frequency significantly contributes to our understanding and identification of gene architecture and functioning.
6. What are the benchmarks for map quality?
Quality determination of maps primarily relies on indicators such as statistical mapping, collinearity analysis of genetic and physical maps, and recombination heatmap analysis of neighboring markers.
7. How does a genetic linkage map differ from a physical map?
A genetic linkage map is organized according to the recombination frequencies, demonstrating the relative positions of genes or markers on a chromosome. In contrast, a physical map is constructed based on the precise physical locations and definitive distances of genes on chromosomes, determined through the actual DNA sequence. The integration of these two types of mapping offers a comprehensive understanding of genomic structure and function.
Genetic Linkage Map Case Studies
An ultra-high-density genetic map provides insights into genome synteny, recombination landscape and taproot skin colour in radish (Raphanus sativus L.)
Journal: Plant biotechnology journal
Impact factor: 13.263
Published: 20 June 2019
Background
High-density genetic maps are essential for genetic and genomic research, including mapping quantitative trait loci (QTLs) in crops. The resolution of QTL mapping depends on marker density and population size, and high-throughput resequencing enhances marker development and genotyping. Anthocyanins, responsible for the colors in various plants, offer health benefits and play roles in attracting pollinators and protection. Meiotic recombination, crucial for creating new allele combinations, affects genomic evolution and plant breeding. High-density genetic maps help study recombination distribution and hotspots. In radish, a high-density linkage map was created, identifying QTLs and candidate genes for important traits, providing insights for genetic improvement and genome evolution studies.
Methods
- Plant materials
- Phenotype data collection
- Two advanced radish inbred lines
- Population resequencing
- Genotyping
- Illumina HiSeq 2500 platform
- Phenotype evaluation
- Bin map construction
- QTL mapping
- RT-qPCR analysis
Results
To construct a high-resolution genetic linkage map in radish, whole-genome resequencing of 137 F2 individuals and their parental lines was performed using the Illumina HiSeqTM 2500 platform. This generated 403 Gb of clean reads, mapped to the radish reference genome, and identified 411,891 SNPs after filtering. Using a sliding window approach, 2,852 recombination bin markers were created, covering a genetic distance of 1,306.8 cM with an average distance of 0.46 cM between markers. Significant segregation distortion was observed in some markers, identifying 19 segregation distortion regions across seven linkage groups. Distorted markers were retained to improve linkage group coverage.
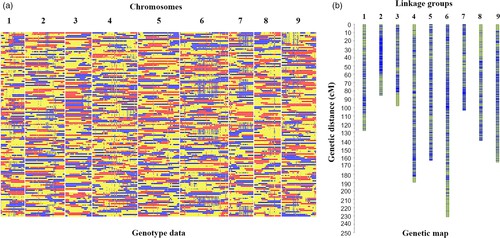 Fig 1. Recombination bin map (a) and genetic map (b) of 137 F2 individuals.
Fig 1. Recombination bin map (a) and genetic map (b) of 137 F2 individuals.
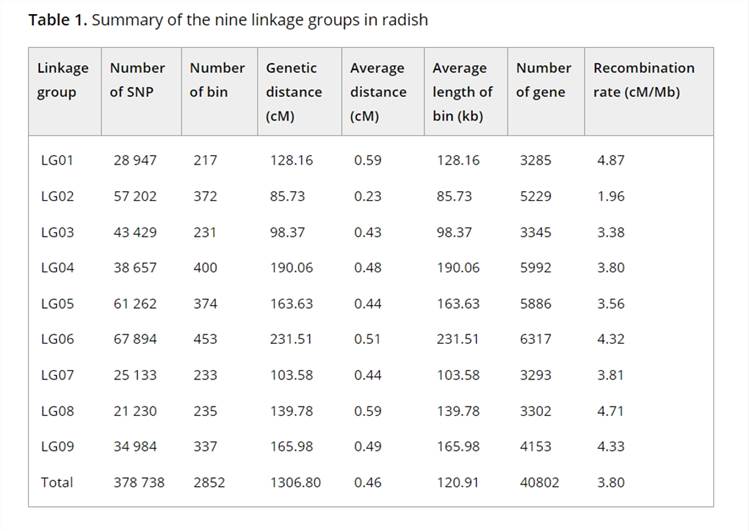
In the F2 population, the detection of 17 quantitative trait loci (QTLs) was ascertained, out of which seven corresponded with antecedently acknowledged QTLs in the same linkage group. The demarcated QTL regions averaged a length of 168 kilobases, crossing five different linkage groups. The method undertaken to isolate the gene influencing root skin color involved a cross between the varieties 'NAU-LB' and 'NAU-YH'. The findings suggested that the dominance of the so-called R gene is responsible for regulating the red skin color. The specific localization of the R gene was pinpointed to a 72 kilobase region situated on chromosome 7. Further investigation identified the RsMYB90 gene as the probable gene implicated in the control of red skin color. It displayed elevated expression levels predominantly in the red-skinned cultivars. Intriguingly, the RsMYB90 gene exhibits a noteworthy degree of homology with members of the MYB transcription factor family, a group renowned for their involvement in the process of anthocyanin biosynthesis.
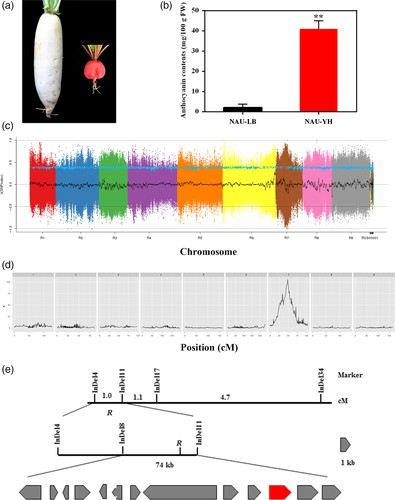 Fig 2. Map-based cloning of red skin gene R.
Fig 2. Map-based cloning of red skin gene R.
The locations of bin markers on the genetic map were compared to their physical positions in the radish genome assembly. All bin markers were mapped onto the nine pseudo-chromosomes, covering 80.7% of the 424 Mb radish reference genome. While there was a high degree of collinearity between the genetic map and the chromosomes, some inconsistencies were noted. Specifically, bins at 26–43 Mb on chromosome 2 were inverted, and the order of bins at the distal ends of chromosomes 1, 3, and 4 did not match the genetic map.
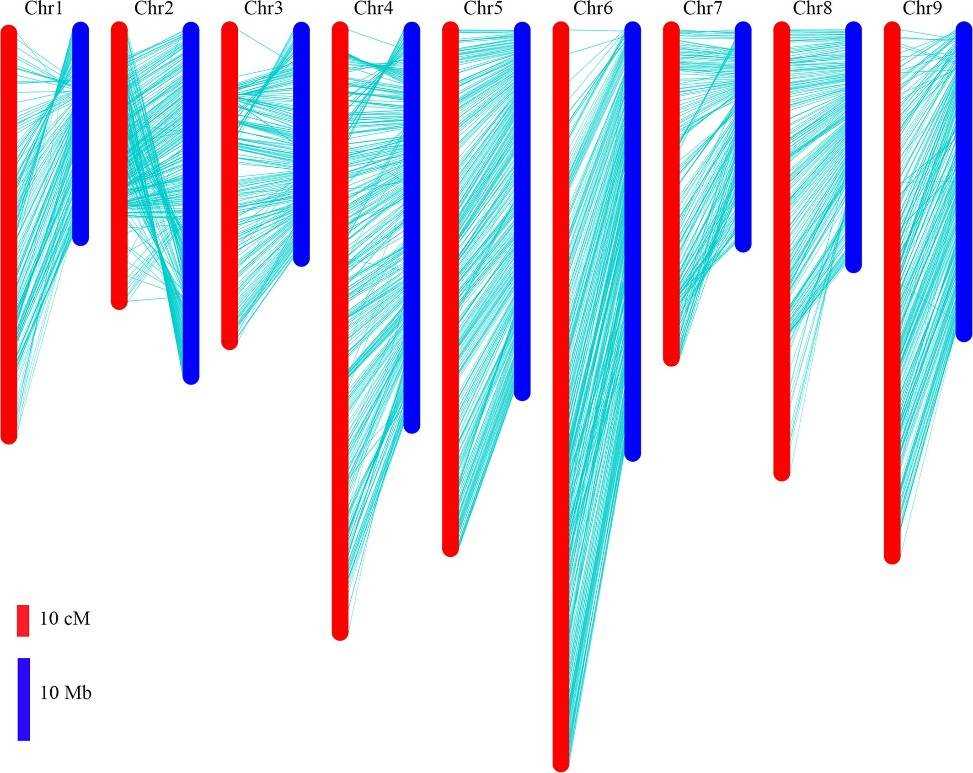 Fig 3. Comparison between the genetic map and radish genome sequence.
Fig 3. Comparison between the genetic map and radish genome sequence.
Conclusion
Through whole-genome sequencing, we have constructed a high-resolution, high-precision dense genetic map. Combined with Quantitative Trait Locus (QTL) mapping and positional cloning, RsMYB90 was identified as a candidate gene controlling the red skin of radish taproots. The RsMYB90 gene plays a crucial role in the regulation of anthocyanin biosynthesis, lending insights into the genetic regulation of anthocyanin accumulation in radishes.
Reference:
- Luo X, Xu L, Wang Y, et al. An ultra-high-density genetic map provides insights into genome synteny, recombination landscape and taproot skin colour in radish (Raphanus sativus L.). Plant biotechnology journal, 2020, 18(1): 274-286.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Collection of genetic data in ethnic-based studies across Aymaras, Quechuas and Mestizos: the challenges of the Genetics of Alzheimer's in Peruvian Population (GAPP) study
Journal: Alzheimer's & Dementia
Year: 2022
Evaluation of Plasma Biomarkers for A/T/N Classification of Alzheimer Disease Among Adults of Caribbean Hispanic Ethnicity
Journal: JAMA Network Open
Year: 2023
Increased Production of Pathogenic, Airborne Fungal Spores upon Exposure of a Soil Mycobiota to Chlorinated Aromatic Hydrocarbon Pollutants
Journal: Microbiology Spectrum
Year: 2023
A Splice Variant in SLC16A8 Gene Leads to Lactate Transport Deficit in Human iPS Cell-Derived Retinal Pigment Epithelial Cells
Journal: Cells
Year: 2021
See more articles published by our clients.


