
 Sample Submission Guidelines
Sample Submission Guidelines
Targeted Region Sequencing
CD Genomics has been providing the fast and affordable targeted region sequencing service for decades. We employ the state-of-the-art sequencing instruments to help you obtain full DNA information of the selected targets in a cost-effective manner, which can greatly increase both the breadth and the depth of your genomics research.
The Introduction of Targeted Region Sequencing
Targeted region sequencing is an effective approach for investigating your selected region(s) of interest by next generation sequencing. By utilizing targeted region sequencing panels, you can discover single-nucleotide polymorphisms (SNPs), insertions/deletions (InDels), copy number variations (CNVs), and structural variants (SVs). Compared with whole genome sequencing, targeted region sequencing enables accurate detection of rare variants with higher sensitivity and specificity. This approach is very cost-effective when handling a large number of samples, which significantly reduces the cost per sample.
The process of targeted region sequencing includes probes/primers designing/synthesis, target regions capture, library construction, paired-end sequencing, and bioinformatics analysis based on target sequences. Specific probe/primer sets are designed to enrich targeted regions using either hybridization or amplification methods. In the hybrid capture method, biotinylated oligonucleotide probes are designed to target regions of interest within a DNA fragment library. After a hybridization incubation, streptavidin-coated magnetic beads are used to capture the biotinylated probe/target hybrids, which results in a library that is highly enriched for the targeted DNA. The current amplification method relies on multiplex PCR or some form of primer extension across regions of interest.
The targeted region sequencing has a wide range of applications, including:
- Detection of SNPs/InDels/CNVs/SVs
- Discovery of germline or somatic mutations
- Detection and quantification of rare variants and low-frequency alleles
- Linkage analysis for inherited diseases
- Discovery of biomarkers and therapeutic targets
Advantages of Targeted Region Sequencing
- Identification of variants at low allele frequencies (down to 1%).
- A smaller data set for bioinformatics analysis.
- Much lower cost with a large number of samples.
- High depth (500-1000X, or higher), allowing identification of rare variants.
Targeted Region Sequencing Workflow
CD Genomics employs Illumina HiSeq instruments to provide the fast and accurate targeted region sequencing and bioinformatics analysis. Our highly experienced experts execute quality management, following every procedure to ensure high quality results. The general workflow for targeted region sequencing is outlined below.

Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategies
|
 |
Bioinformatics Analysis We provide customized bioinformatics analysis including:
|
Analysis Pipeline
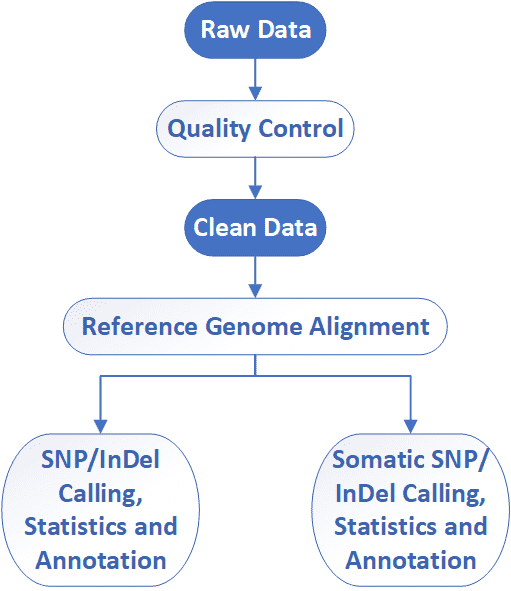
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Targeted Region Sequencing for your writing (customization)
CD Genomics provides full targeted region sequencing service package including sample standardization, probe/primer designing, targeted capture, library construction, deep sequencing, raw data quality control and bioinformatics analysis. We can tailor this pipeline to your research interest. If you have additional requirements or questions, please feel free to contact us.
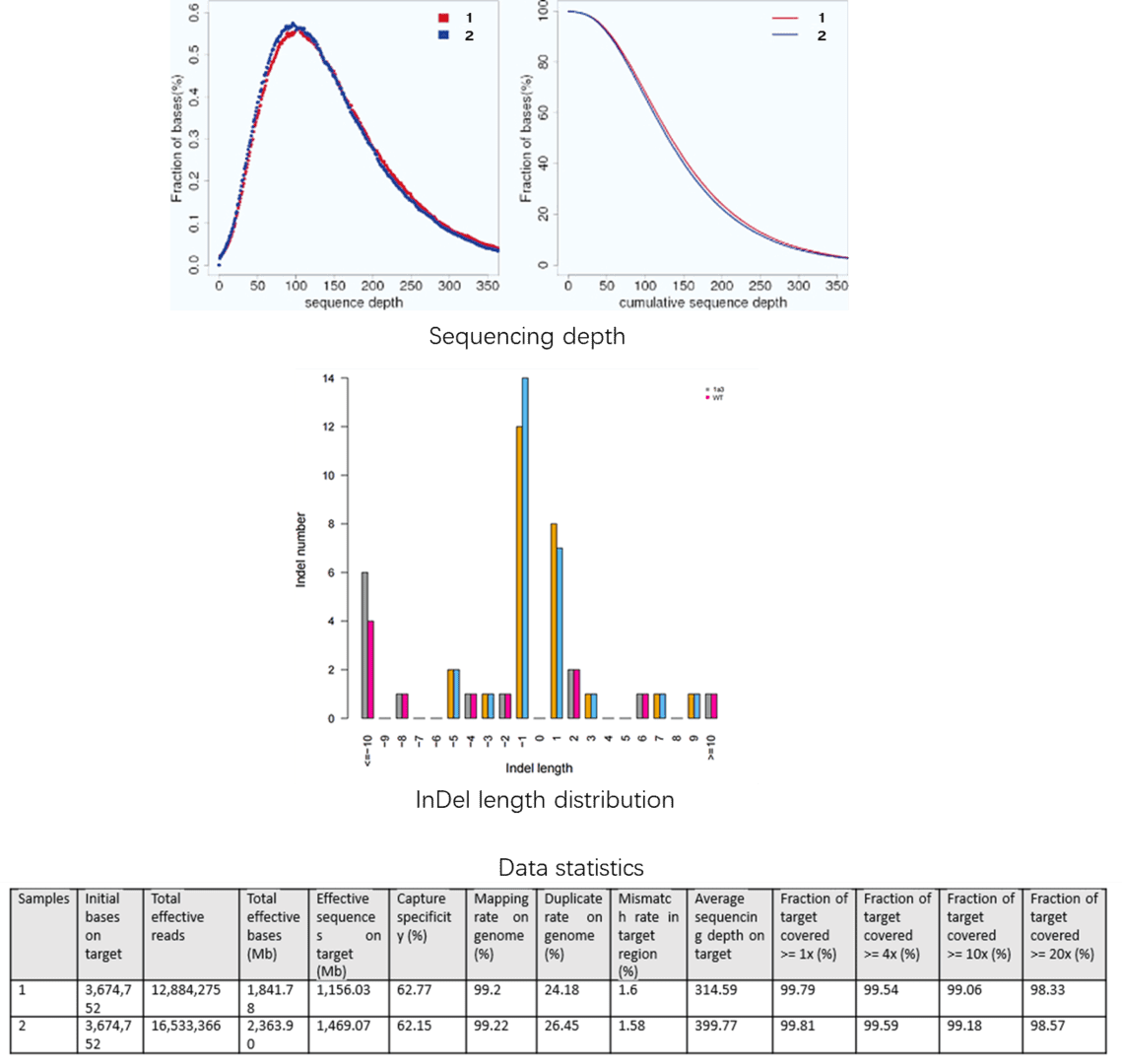
Demo Results
Targeted Region Seq FAQs
1. What is the difference between whole exome sequencing and targeted region sequencing?
In spite of probably the same principle and workflow, whole exome sequencing focuses on exome while targeted region sequencing focuses on any genes that are defined by you.
2. What do I need to submit?
The customer who requires the targeted region service need submit the DNA or tissue samples, as well as the list of gene candidates or the chromosomal localization of your target region. We are responsible for the probes generation, target enrichment, library construction, sequencing, and bioinformatics analysis.
We accept the following sample types for targeted region sequencing: purified genomic DNA, PCR amplicons, frozen cell pellets, bacterial colonies, FFPE (formalin-fixed, paraffin-embedded) tissue sections, tissues, blood, and swabs.
3. How about the turnaround time?
The targeted region sequencing requires custom-made probes, and the custom-made probe need further optimization for an effective enrichment of target regions. It often takes 40 working days to complete the customization of probes and the deep sequencing. But there are commercial solutions for targeted region sequencing, such as the SeqCap EZ Prime Choice Probes, which can eliminate the process of the customization of probes.
4. How to validate variants?
Variants identified through the next-generation sequencing (NGS) require further confirmation using either NGS or Sanger sequencing. According to the survey did by Coppieters in September 2014, almost 70% of 178 respondents indicated that they are currently validating their NGS findings using NGS or Sanger sequencing. We will validate variants using NGS and Sanger sequencing.
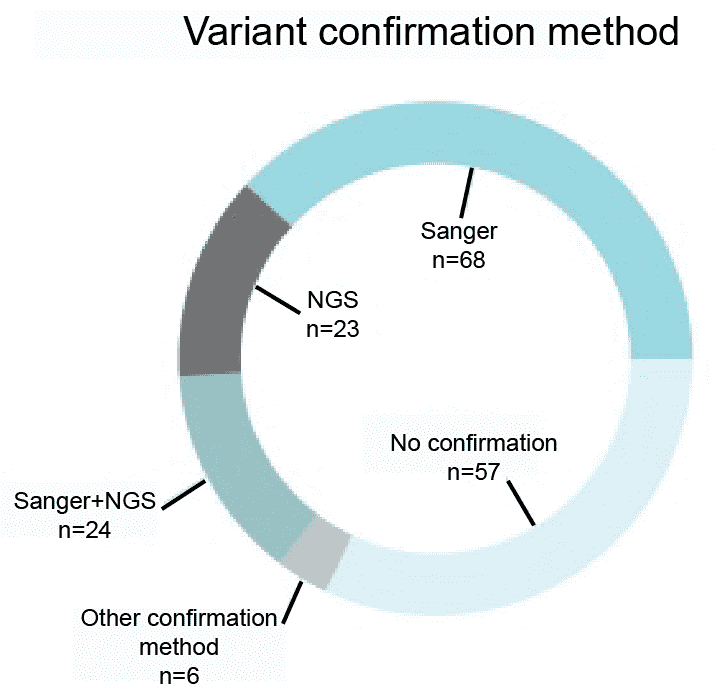 Figure 1. A survey of the variant confirmation methods (Coppieters et al. 2016).
Figure 1. A survey of the variant confirmation methods (Coppieters et al. 2016).
Reference:
- Coppieters F, Verniers K, De Leeneer K, et al. Targeted resequencing and variant validation using pxlence PCR assays. Biomolecular detection and quantification, 2016, 6: 22-26.
Targeted Region Seq Case Studies
Identification of a novel missense mutation of MIP in a Chinese family with congenital cataracts by target region capture sequencing
Journal: Scientific Reports
Impact factor: 4.259
Published: 06 January 2017
Abstract
The authors recruited a Chinese family with autosomal dominant congenital cataracts (ADCC). They found a heterozygous missense mutation c.634G>C(p.G212R) substitution in the MIP gene by using target region sequencing. In conclusion, the study presented genetic and functional evidence for this novel mutation, which leads to congenital progressive cortical punctate with or without Y suture. The proband (II:2) had a different type of
Results
1. Clinical features
The authors investigated a Chinese family (Figure 1), and found that the proband (II:2) showed a punctate cataract, while the other affected members show punctate cortical opacities, combined with Y-sutural cataracts (Figure 2).
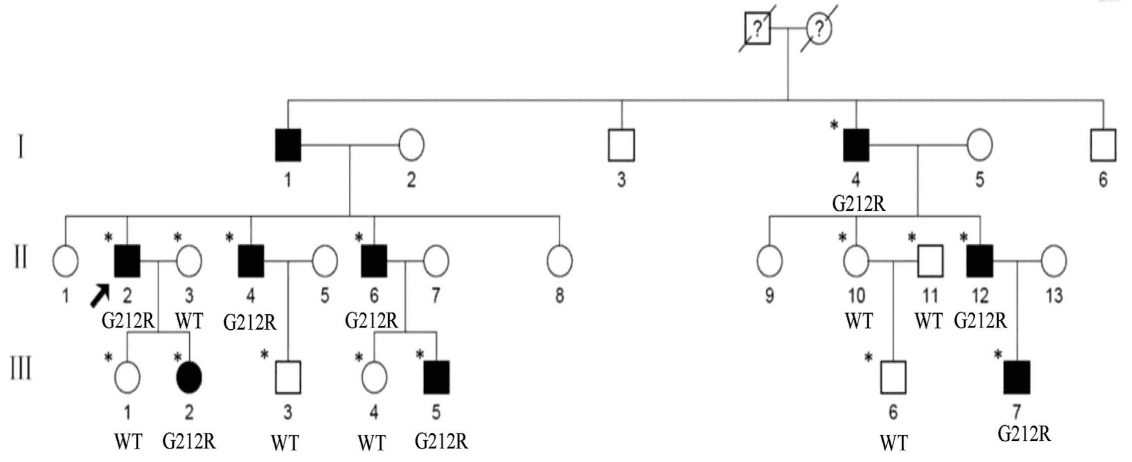 Figure 1. Pedigree of the family. Squares and circles denote males and females, respectively. Black symbols
denote affected members, while open symbols denote unaffected individuals. The diagonal line denotes a decreased
family member, and the black arrow denotes the proband. Asterisks denote sequenced individuals.
Figure 1. Pedigree of the family. Squares and circles denote males and females, respectively. Black symbols
denote affected members, while open symbols denote unaffected individuals. The diagonal line denotes a decreased
family member, and the black arrow denotes the proband. Asterisks denote sequenced individuals.
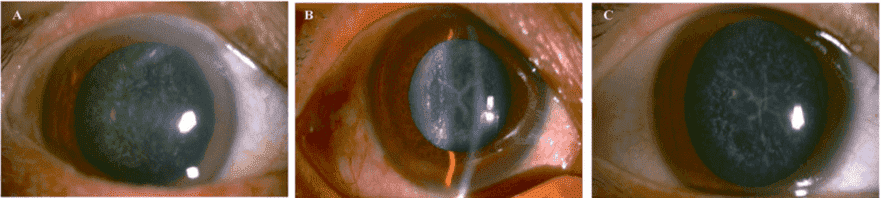 Figure 2. Slit lamp photographs of the patients. The proband II:2 (A) showed a punctate cataract, while his
younger brother II:6 (B) and the brother's son III:5 (C) showed a Y-suture cataracts combined with punctate
cortical opacities.
Figure 2. Slit lamp photographs of the patients. The proband II:2 (A) showed a punctate cataract, while his
younger brother II:6 (B) and the brother's son III:5 (C) showed a Y-suture cataracts combined with punctate
cortical opacities.
2. Target region sequencing and variant analysis
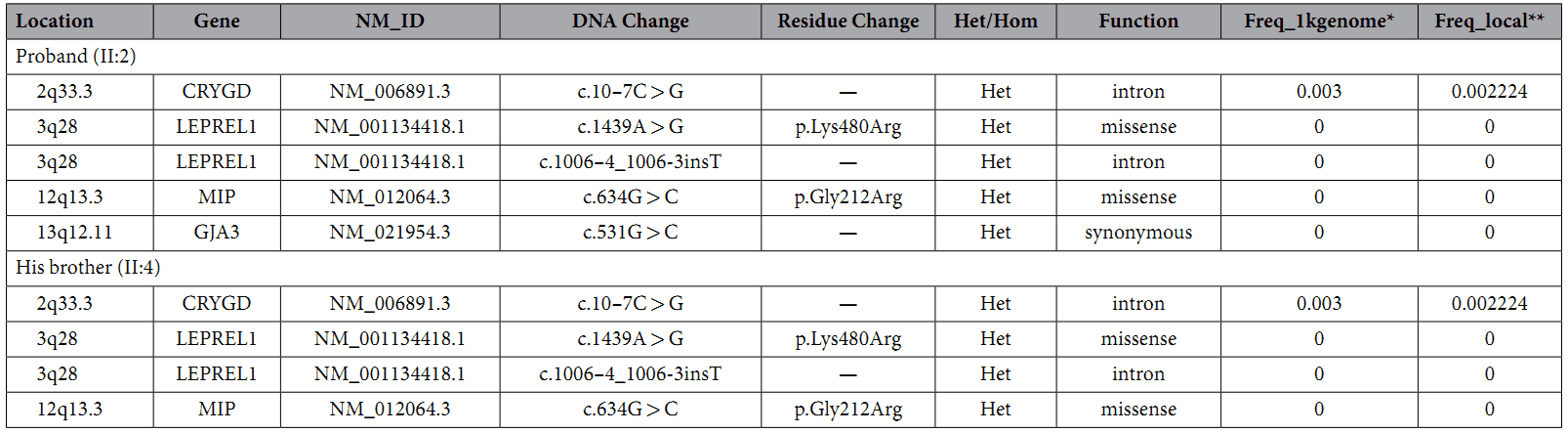
A custom-made capture panel was designed to capture 351 genetic eye disease genes that are collected from OMIM (Human Genetics Knowledge for the World) and the published literatures. The authors found four rare variants in both samples (Table 1).
Table 1. Rare variants in congenital cataract causing genes.
3. c.634G>C mutation
Only the c.634G>C mutation of the MIP gene was a novel heterozygous missense mutation, which changed Glycine to Arginine at the position 212 (p.G212R). And segregation analysis was revealed that the mutation cosegrageted well with all affected participants and was not detected in unaffected members or the 100 normal controls.
PolyPhen-2 and SIFT analysis strongly indicated that the p.G212R mutation is likely to be deleterious to the protein and may be responsible for the congenital cataracts. A multiple sequence alignment showed that the Glycine at position 212 of MIP is highly conserved among various species (Figure 3B).
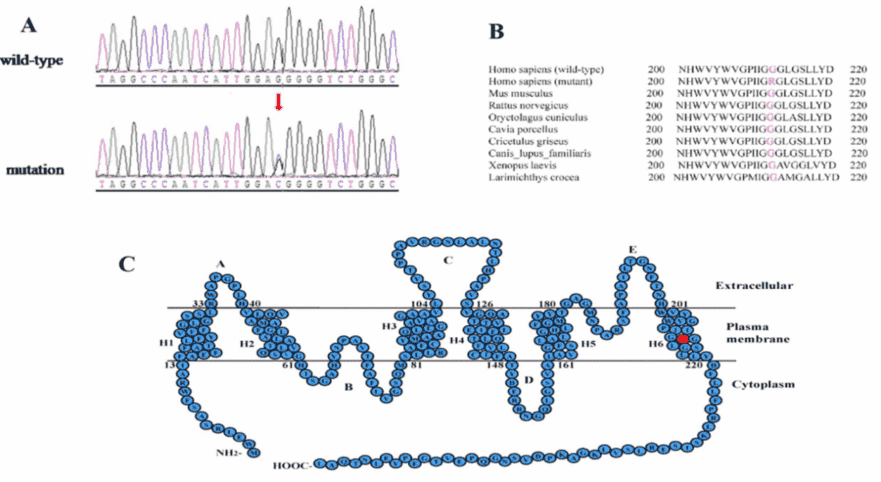 Figure 3. mutation analysis of the MIP gene.
Figure 3. mutation analysis of the MIP gene.
mRNA and protein levels of WT-MIP and G212R-MIP
The authors investigated the expression of wide-type MIP and G212R-MIP in transfected Hela cells. It showed that the green fluorescence in Hela cells transfected with the G212R construct was much weaker than those transfected with the wide-type construct.
The RT-PCR was used to measure the RNA transcription level of WT-MIP and G212R-MIP, which revealed that a similar relative MIP mRNA expression levels (Figure 4B). Nevertheless, Western blot indicated that the protein expression levels of MIP were significantly reduced in the G212R-MIP cells (Figure 4C) in accord with the results of fluorescence microscopy analysis.
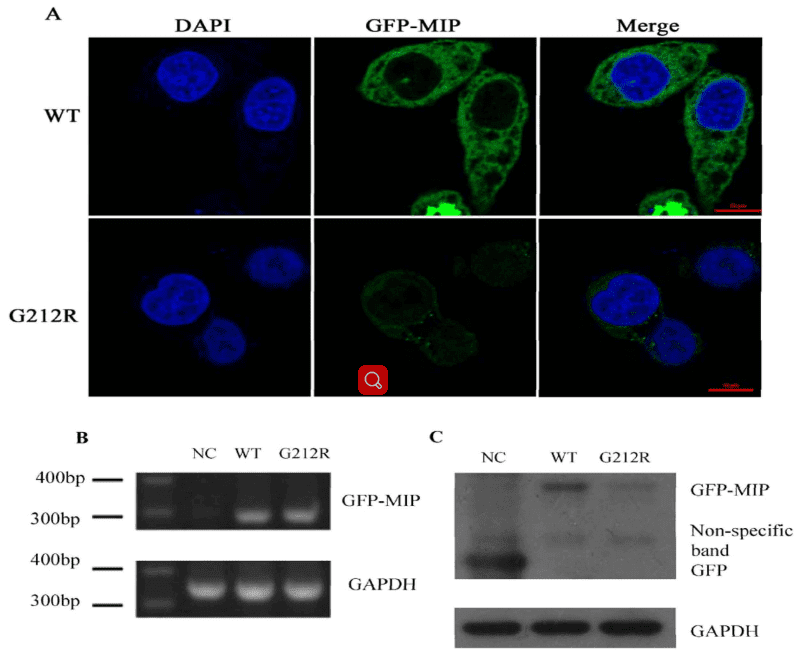 Figure 4. mRNA and protein levels of WT-MIP and G212R-MIP.
Figure 4. mRNA and protein levels of WT-MIP and G212R-MIP.
Conclusions
In the present study, the authors suggest that the c.634G>C, G212R substitution may contribute to the pathogenesis of ADCC in this Chinese family.
Target region sequencing is a powerful tool for clinical diagnosis of a heterogeneous group of monogenic disorders due to the compatible performance in capture coverage, the lower cost, and the shorter turnaround time.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Genetic variants in the bipolar disorder risk locus SYNE1 that affect CPG2 expression and protein function
Journal: Molecular Psychiatry
Year: 2019
High-Density Mapping and Candidate Gene Analysis of Pl18 and Pl20 in Sunflower by Whole-Genome Resequencing
Journal: International Journal of Molecular Sciences
Year: 2020
Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI
Journal: New phytologist
Year: 2017
Generation of a highly attenuated strain of Pseudomonas aeruginosa for commercial production of alginate
Journal: Microbial Biotechnology
Year: 2019
Combinations of Bacteriophage Are Efficacious against Multidrug-Resistant Pseudomonas aeruginosa and Enhance Sensitivity to Carbapenem Antibiotics
Journal: Viruses
Year: 2024
Genome Analysis and Replication Studies of the African Green Monkey Simian Foamy Virus Serotype 3 Strain FV2014
Journal: Viruses
Year: 2020
See more articles published by our clients.


