
 Sample Submission Guidelines
Sample Submission Guidelines
CRISPR Sequencing
CD Genomics provides CRISPR-Cas9 knockout validation and potential off-target detection in a high-throughput and cost-effective manner by harnessing advanced next generation sequencing (NGS). Our team members have experience in both genome editing and NGS, enabling extensive support to your research.
What is CRISPR
CRISPR, short for Clustered Regularly Interspaced Short Palindromic Repeats, is a revolutionary gene-editing technology originating from the natural immune systems of bacteria and archaea. This system is a crucial component of prokaryotic immune defense, enabling these organisms to capture fragments of viral DNA and use them to detect and destroy similar viral sequences during subsequent infections. In essence, CRISPR allows viruses to integrate their genetic material into bacteria, leveraging bacterial cellular machinery to replicate their own genes, a process we refer to as gene editing.
The primary components of the CRISPR system include:
- CRISPR Loci: These loci consist of repetitive genetic sequences interspersed with "spacers," which are fragments of viral DNA captured from previous infections.
- Cas Proteins (CRISPR-associated proteins): Cas9, for example, is an enzyme that can cut DNA at specific locations, guided by RNA sequences.
- Guide RNA (gRNA): This is a short RNA sequence complementary to the target DNA sequence, directing the Cas9 enzyme to the precise site in the genome where editing is desired.
What is CRISPR-Cas9 System
The CRISPR-Cas9 gene targeting system, requiring two components: a custom guide RNA (sgRNA, consisting of target-specific crRNA sequence and tracrRNA) and a non-specific CRISPR-associated endonuclease (Cas9), is a new genome engineering tool that enables researchers to edit parts of the genome by removing, adding or altering a section of the DNA sequence in organism. It is currently the simplest, most versatile, precise and effective method of genetic manipulation that has many potential applications including medicine and crop seed enhancement. CRISPR-Cas9 has also been adapted to enable high-throughput genome editing and has revolutionized the generation of targeted mutations.
Introduction to CRISPR Sequencing
CRISPR Sequencing is an advanced methodology that integrates CRISPR gene-editing technology with high-throughput sequencing, aimed at precisely manipulating the genome and analyzing the resultant effects. CRISPR, an acronym for Clustered Regularly Interspaced Short Palindromic Repeats, derives from a bacterial immune mechanism and employs Cas (CRISPR-associated) proteins guided by specific RNA molecules to target and edit DNA sequences. This technique not only allows for targeted genome modifications but also enables comprehensive downstream analysis, offering significant potential in both basic research and applied biotechnology.
In CRISPR experiments, the validation of genetic edits is paramount. NGS offers a straightforward, high-throughput approach for the examination of desired mutations, given the potential occurrence of off-target modifications instead of the intended gene edits. CRISPR amplicon sequencing has become the standardized validation technique across academic, clinical, and industrial domains. High-throughput CRISPR screening leverages the principles of targeted amplicon sequencing by employing primers flanking the target regions for PCR amplification. Targeted amplicon sequencing stands as the most sensitive method for mutation detection, capable of identifying mutations with frequencies as low as 0.01%.
To address the emerging needs of research communities, CD Genomics has developed an affordable, reliable, and high-throughput strategy for screening and validating CRISPR-Cas9 based mutations by harnessing amplicon based next-generation Sequencing. Our CRISPR next-generation sequencing service can give you direct and detailed information about the nature and diversity of the mutations, including confirmation of knockout/knockin alleles, assessment of sgRNAs cutting efficiency, homozygous and heterozygous identification, mutation frequencies calculation et al.
Advantages of Our CRISPR Sequencing Service
- The high precision of the CRISPR/Cas9 system allows for accurate targeting and editing of specific genomic loci
- This versatility facilitates a variety of genomic modifications, including gene knockouts, knock-ins, point mutations, and gene regulation
- Extensive multiplexing flexibility and high-throughput sequencing, up to 104 samples per run
- Ultra-deep sequencing of amplicons or captured regions, in excess of 1000X coverage
- Cost-effective, and highly sensitive detection levels without bias
- No need for laborious and time-consuming cloning steps
- Dedicated support from specialized Ph.D.-level scientists
Applications of CRISPR Sequencing
- Functional Genomics: This field investigates gene function and the roles genes play in biological processes, providing a comprehensive understanding of genomic contributions to various cellular activities.
- Gene Therapy: Focusing on rectifying pathogenic mutations responsible for hereditary diseases, gene therapy aims to restore normal gene function and alleviate disease symptoms.
- Agricultural Biotechnology: This discipline involves the enhancement of crop traits, such as disease resistance and yield, through genetic modifications, thereby contributing to agricultural productivity and sustainability.
- Cancer Research: Dedicated to elucidating the roles of tumor-related genes, cancer research explores the mechanisms of oncogenesis and cancer progression, paving the way for novel therapeutic strategies.
CRISPR Sequencing Workflow
Our highly experienced expert team executes quality management following every procedure to ensure comprehensive and accurate results. Our CRISPR mutation sequencing workflow is outlined below, including DNA isolation, PCR experiments, deep sequencing, and data analysis. Our CRISPR mutation sequencing enables researches to validate guides library, validate CRISPR/Cas9 targets and mutation efficiency, as well as discover the most promising target sites.

Service Specifications
Sample Requirements
|
|
 |
Sequencing Strategy
|
 |
Bioinformatics Analysis
We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
Genome Editing with CRISPR: How to Effectively Minimize Off-Target Effects
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in CRISPR Sequencing for your writing (customization)
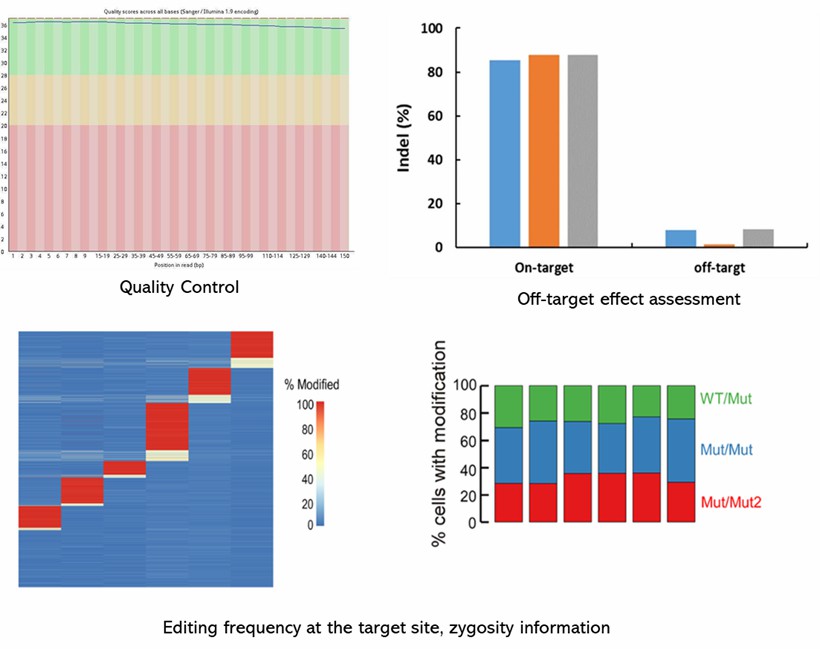
CD Genomics offers comprehensive services to detect potential off-target effects through targeted enrichment and deep sequencing, all at competitive prices. Our advanced capabilities allow us to sequence hundreds of samples concurrently, ensuring efficient processing and high throughput. The distribution patterns of insertions and deletions (InDels) will be meticulously analyzed, and mutations will be verified using the robust CRISPResso tool. Should you have any further requirements or inquiries, please feel free to reach out to us.
Demo Results
Partial results are shown below:
CRISPR Seq FAQs
1. Is CRISPR-Cas9 next generation sequencing?
Next-generation sequencing (NGS) plays an integral role in various stages of CRISPR-Cas9 genome editing workflows. Its applications range from comprehensive genome sequencing to analyze potential off-target effects of CRISPR, to targeted sequencing aimed at confirming specific gene knockouts and other edits. By employing NGS, researchers can achieve detailed and precise validation of CRISPR-induced modifications, ensuring the accuracy and reliability of their genetic alterations.
2. What are the ethical considerations of CRISPR Sequencing?
CRISPR sequencing raises ethical concerns, particularly regarding the potential for off-target mutations and the implications of germline editing. Ethical guidelines emphasize the need for rigorous safety assessments, informed consent, and transparency in research involving gene editing technologies.
3. How do researchers ensure the specificity of CRISPR Sequencing results?
Researchers employ bioinformatics tools to analyze sequencing data and distinguish genuine CRISPR-induced mutations from background noise or sequencing errors. Additionally, validation techniques such as Sanger sequencing or droplet digital PCR (ddPCR) can confirm the presence of mutations at specific genomic loci.
4. How is CRISPR Sequencing used in clinical research and therapy?
In the domain of clinical research, CRISPR sequencing plays an integral role in the advancement of therapeutic prospects for genetic disorders. This technology facilitates the precise validation of genomic modifications in patient-derived cells, thereby allowing researchers to thoroughly investigate the efficacy and safety profiles of gene therapies ahead of clinical trials. Moreover, CRISPR sequencing is instrumental in elucidating disease mechanisms and pinpointing potential therapeutic targets, fostering a deeper understanding that may drive the development of innovative treatment strategies.
CRISPR Seq Case Studies
Genome-wide off-target analyses of CRISPR/Cas9-mediatedT-cell receptor engineering in primary human T cells
Journal: Clinical & Translational Immunology
Impact factor: 5.8
Published: 23 January 2022
Background
The utilization of T cells for the defense against cancer is a prominent approach in contemporary immunotherapeutic strategies. Genetic engineering of T-cell receptors (TCRs) enables the redirection of T cell specificity, the elimination of alloreactivity, and the advancement of adoptive T-cell transfer (ACT) therapy. The introduction of DNA double-strand breaks via CRISPR/Cas9 technology facilitates gene knockout or knock-in manipulations. An effective method for ascertaining the safety of engineered T cells involves the detection of off-target genes across the entire genome. Leveraging CRISPR/Cas9 techniques, the authors employed ribonucleoprotein delivery to knock out the TCR, allowing them to assess the safety of genetically engineered T cells. Whole-genome sequencing was subsequently employed to investigate whether CRISPR/Cas9-mediated double-strand breaks at TCR sites are associated with off-target effects in primary T cells.
Materials & Methods
Sample Preparation
- Human
- T cell
- TCR knockout
Sequencing
- Whole-Genome Sequencing (WGS)
- Targeted deep sequencing
- Analysis of Cas9 off-target sites
- Analysis of predicted Cas9 off-target sites
- Analysis of electroporation-dependent increase in mutation rate
Results
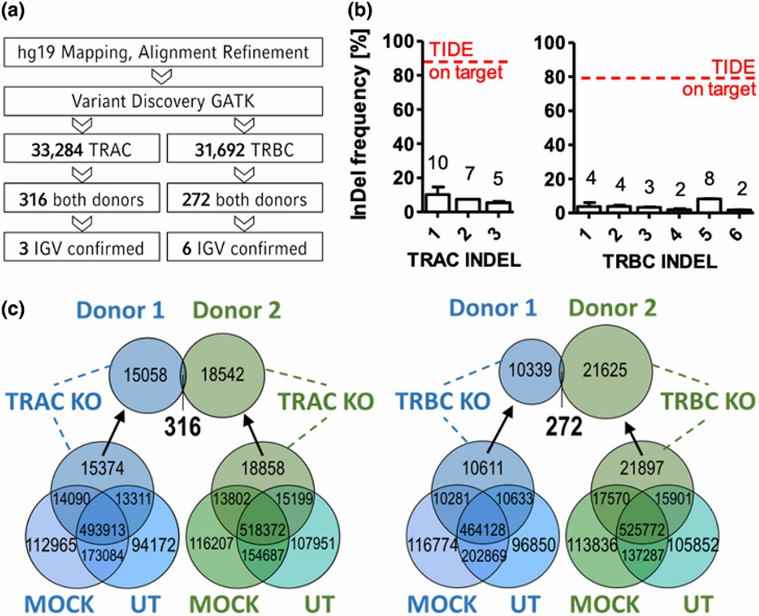 Fig 1. Whole-genome analysis of gRNA-dependent Cas9-induced off-target effects.
Fig 1. Whole-genome analysis of gRNA-dependent Cas9-induced off-target effects.
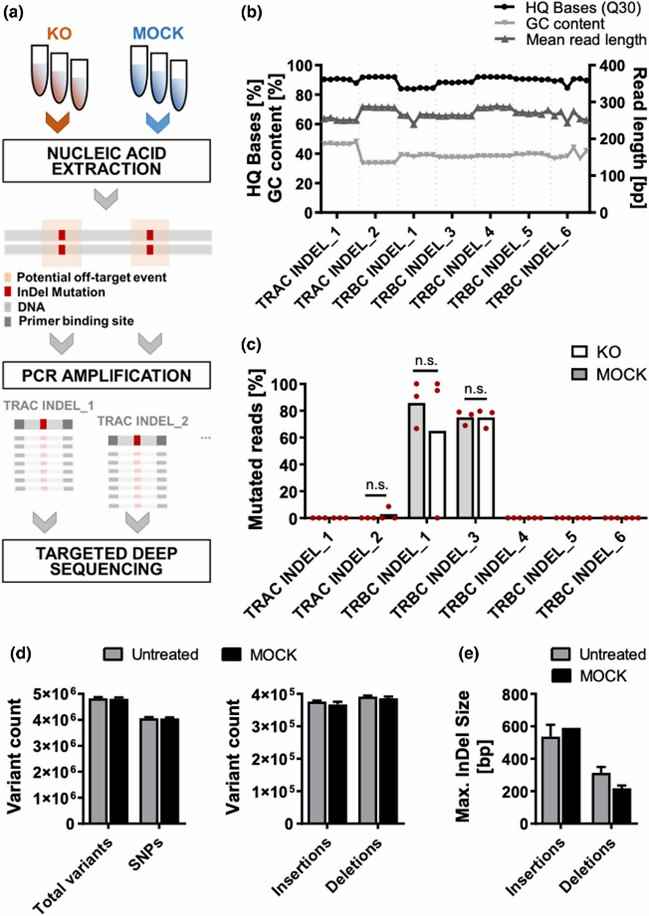 Fig 2. Targeted deep sequencing of WGS-pre-identified off-target events.
Fig 2. Targeted deep sequencing of WGS-pre-identified off-target events.
The safety of T cells was assessed by examining nonspecific nuclease-induced off-target incidents. Utilizing Cas-OFFinder, the authors screened for matching sites with a mismatch of ≤ 4 between the guide RNA (gRNA) and non-target DNA, considering them as possible off-target locations. Subsequently, whole-genome sequencing was carried out on both untreated samples and MOCK samples. The results revealed that electroporation alone did not yield any significant genomic variations.
The authors then conducted whole-genome sequencing on samples subjected to TRAC and TRBC edits, excluding the InDels found in untreated and MOCK samples. Employing a whole-genome homologous sequence alignment based on the single-guide RNA (sgRNA), they identified 316 distinct InDels in the TRAC-edited samples and 272 distinct InDels in the TRBC-edited samples. These potential off-target sites were subsequently compiled by the authors.
Conclusion
In summary, this study investigates the safety of CRISPR/Cas9-mediated genetic engineering in T-cell receptors (TCR), aiming to assess off-target effects. Using a combined approach of biased target prediction, unbiased whole-genome sequencing, and targeted deep sequencing, the study confirmed high efficiency in TCR knockout without detecting any CRISPR/Cas9-induced off-target InDel mutations. However, further in vivo research is necessary to fully evaluate the clinical safety of genetically modified T-cell products.
Reference
- Kaeuferle T, Stief T A, Canzar S, et al. Genome‐wide off‐target analyses of CRISPR/Cas9‐mediated T‐cell receptor engineering in primary human T cells. Clinical & Translational Immunology, 2022, 11(1): e1372.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
The HLA class I immunopeptidomes of AAV capsid proteins
Journal: Frontiers in Immunology
Year: 2023
Isolation and characterization of new human carrier peptides from two important vaccine immunogens
Journal: Vaccine
Year: 2020
Change in Weight, BMI, and Body Composition in a Population-Based Intervention Versus Genetic-Based Intervention: The NOW Trial
Journal: Obesity
Year: 2020
Sarecycline inhibits protein translation in Cutibacterium acnes 70S ribosome using a two-site mechanism
Journal: Nucleic Acids Research
Year: 2023
Identification of a Gut Commensal That Compromises the Blood Pressure-Lowering Effect of Ester Angiotensin-Converting Enzyme Inhibitors
Journal: Hypertension
Year: 2022
A Splice Variant in SLC16A8 Gene Leads to Lactate Transport Deficit in Human iPS Cell-Derived Retinal Pigment Epithelial Cells
Journal: Cells
Year: 2021
See more articles published by our clients.


