
 Sample Submission Guidelines
Sample Submission Guidelines
Transcriptome Sequencing
CD Genomics is proud of offering and tailoring most types of transcriptomics services to meet the research objectives and budget of our clients. We are able to guide you all the way from project design, project initiation, to high-quality results.
What Is Transcriptome
The transcriptome is the set of all RNA molecules, including mRNA, rRNA, tRNA, and other noncoding RNA, isolated from one cell or a population of cells. Transcriptomics is the high-throughput study of cellular gene expression under specific conditions by cataloging the complete set of RNA transcripts, including mRNA and non-coding RNAs. Transcriptomics enables a genome-wide analysis of transcription at single nucleotide resolution, including determination of the relative abundance of transcripts, unbiased identification of alternative splicing events and post-transcriptional RNA editing events, and detection of single nucleotide polymorphisms (SNPs). This method is advantageous over the existing methods, such as microarrays and expression sequence tags. It is a powerful tool for studies in early embryo development, cellular differentiation, phylogenetic inference, and biomarker discovery.
Transcriptomics Study Procedures
Total RNA is firstly isolated from a sample, then library preparation may involve such steps as poly-A-based mRNAs enrichment or ribosomal-depletion-based RNAs enrichment, RNA fragmentation, reverse transcription to cDNA, adapter ligation, and PCR amplification. Ribosomal depletion is superior to poly-A selection due to its enrichment of RNA with or without poly-A tails. Therefore, transcriptome that has been rRNA depleted is often considered as total RNA, including both mRNA and non-coding RNA. The following deep sequencing is performed with configuration of Illumina HiSeq (single-end, short-read, or paired-end, long-read) or PacBio SMRT (very long-read reaction). The very long-read sequencing strategy is able to sequence the full-length isoforms without the need for assembly. The final step for transcriptomics study is the bioinformatics analysis, generally involving quality trimming of raw data, alignment/mapping to a reference genome or de novo assembly, post-mapping manipulation and quality control, as well as advanced data mining, such as quantification of gene expression.
Our Transcriptomics Services Include:
- RNA-Seq: Reveal RNAs presence and RNAs expression levels in a certain condition and time, enable discovery of novel gene structures, alternatively spliced isoforms, gene fusions, and SNPs/InDels.
- Small RNA Sequencing: Profiles a variety of small non-coding RNAs—miRNAs, siRNAs, piRNAs, snoRNAs, snRNAs, tRNA fragments (tRFs), and more. Detects isoforms, predicts novel sncRNAs, and infers potential targets and functions
- LncRNA Sequencing: Provide the comprehensive analysis for both lncRNAs (the non-coding RNAs longer than 200 nt) and mRNAs at a single sequencing run, enabling the comparison of lncRNAs and mRNAs expressions and function inference of lncRNAs and mRNAs.
- CircRNA Sequencing: Provide the circular RNA (circRNA) sequence information with single-base resolution in one time for potential therapeutic and research applications.
- Degradome Sequencing: Analyze patterns of RNA degradation. Identify miRNA cleavage sites from the degradome effectively and infer target genes accurately.
- Bacterial RNA Sequencing: Provide prokaryotic RNA sequencing service to advance your bacterial gene expression profiling needs by utilizing the latest techniques.
- Ribosome Profiling: Provide a ribosome-profiling strategy that is based on the deep sequencing of ribosome-protected mRNA fragments and enables genome-wide investigation of translation with sub cordon resolution.
- Total RNA-Seq: Analyze both coding and multiple forms of noncoding RNA for a comprehensive view of the transcriptome.
- Targeted RNA-sequencing: Providing qualitative and quantitative information for differential expression analysis, allele-specific expression measurement, and gene fusion verification.
- Exosomal RNA Sequencing: Provide the exosome researchers with a comprehensive service to investigate exosome-associated RNA information.
- ultra-low input RNA sequencing: Allow the study of samples with a limited number of cells or with the ultra low amount of input RNA.
- Dual RNA-seq: Provide direct insight into host–pathogen interplay.
- microRNA Sequencing: Specifically profiles miRNAs—small (~21–23 nt) non-coding RNAs that regulate gene expression post-transcriptionally. Our end-to-end service covers sample QC, library prep, high-throughput sequencing, and data analysis. You'll receive single-base resolution quantification of known miRNAs, detection of novel miRNAs, and insights into miRNA–mRNA regulatory networks. Perfect for biomarker discovery and gene regulation studies.
- mRNA Sequencing Service: Quantifies coding transcriptomes using Illumina NovaSeq. We deliver expression profiling, splice variant detection, gene fusion identification, and RNA editing analysis. Services include QC, library construction, high-throughput sequencing, and advanced bioinformatics tailored to your project goals. Ideal for gene expression research in disease models and pharmacogenomics.
Service Advantages
Detection of All RNA Types: Traditional transcriptome sequencing utilizes the Oligo d(T) method to purify RNA molecules with Poly(A) tails. However, many popular RNA molecules, such as circular RNA and certain long non-coding RNAs (LncRNAs), lack Poly(A) tails, leading to their loss during processing. CD Genomics offers rRNA depletion kits designed for various species, enabling the retention of all RNA types.
Suitable for Degraded Samples: CD Genomics employs proprietary kits that are compatible with degraded samples, allowing for the preservation of RNA information in highly degraded samples. This approach is particularly well-suited for transcriptome analysis of clinical samples.
Strand-Specific Detection: Many genetic loci produce antisense RNAs with crucial regulatory functions, transcribed in the opposite direction. Conventional library preparation methods cannot distinguish these antisense RNAs, but CD Genomics utilizes strand-specific library preparation, enabling the accurate detection of antisense RNAs, thus providing more comprehensive transcriptome information.
Specialized Bioinformatics Analysis: We boasts a robust bioinformatics team capable of meeting various in-depth data analysis requirements for clients.
High Resolution: It can discern similar genes within gene families and single-nucleotide differences caused by variable splicing.
Broad Detection Range: It can precisely count copies from a few to tens of thousands, simultaneously identifying and quantifying both common and rare transcripts.
Data Analysis
Differential mRNA, Circular RNA, and Long Non-Coding RNA (LncRNA) Expression Analysis
Following the acquisition of high-throughput sequencing data, we employ stringent criteria, requiring a minimum Fold Change of ≥ 2 and a significance level with a P-value of ≤ 0.05, to identify differentially expressed mRNA, circular RNA, and LncRNA.
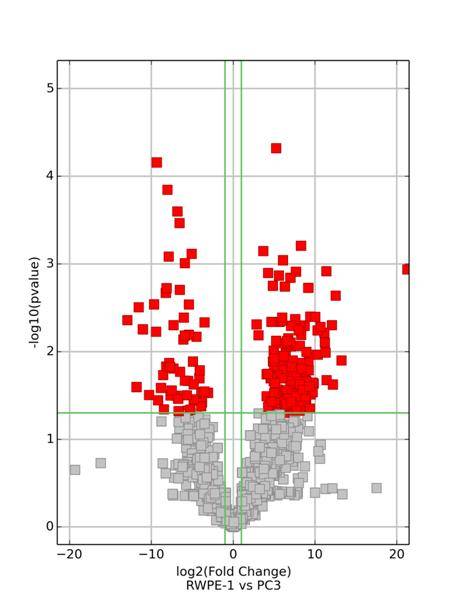
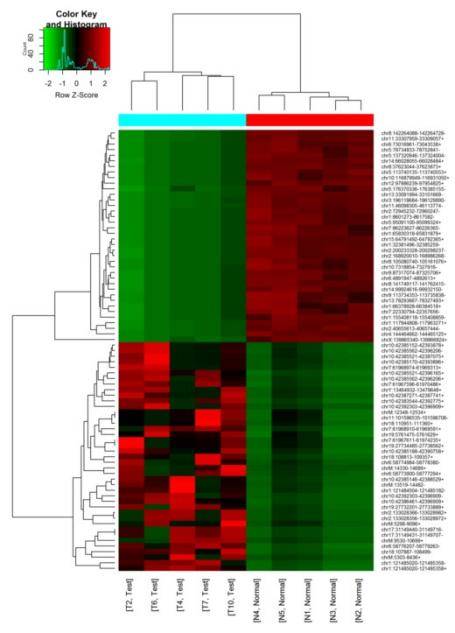
Clustering of Differentially Expressed mRNA, Circular RNA, and LncRNA
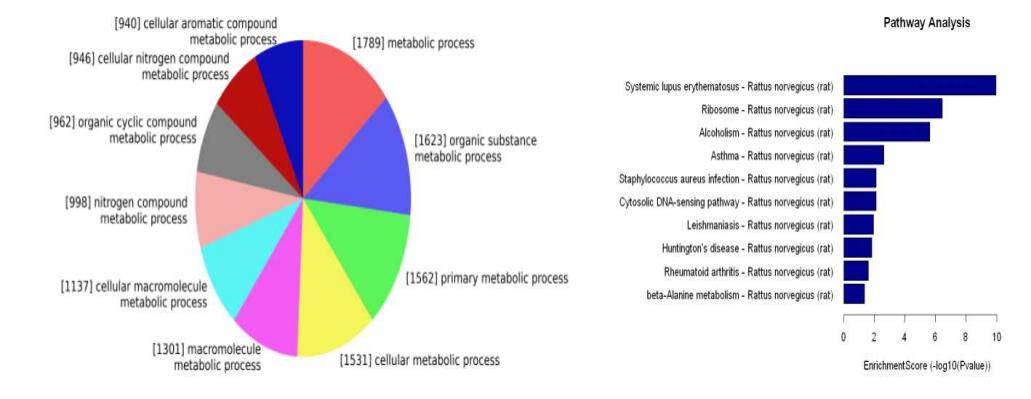
Gene Ontology (GO) and Signaling Pathway Analysis of Differentially Expressed mRNA, Circular RNA, and LncRNA
Conducting functional and signaling pathway enrichment analyses on the source genes of differentially expressed mRNA, circular RNA, and LncRNA facilitates the inference of the functions of these RNA molecules.
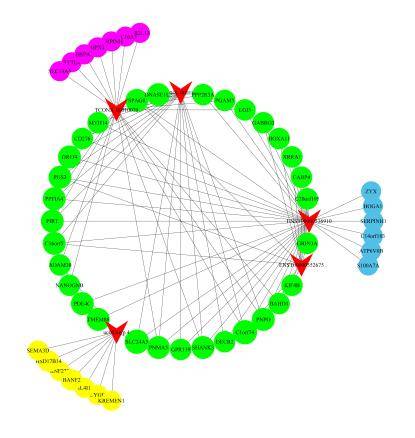
Construction of Long Non-Coding RNA-mRNA Co-expression Network
Leveraging the co-expression association between Long Non-Coding RNA (LncRNA) and messenger RNA (mRNA), we formulate a co-expression network diagram. This approach aids in the identification of LncRNA target genes and the deduction of LncRNA functions.
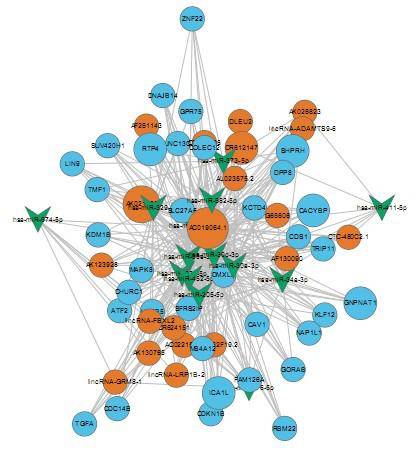
Competitive Endogenous RNA (ceRNA) Analysis
Through the analysis of the circular RNA-microRNA interaction network, we can aid in unraveling the function and mechanisms of circular RNAs acting as miRNA sponges.
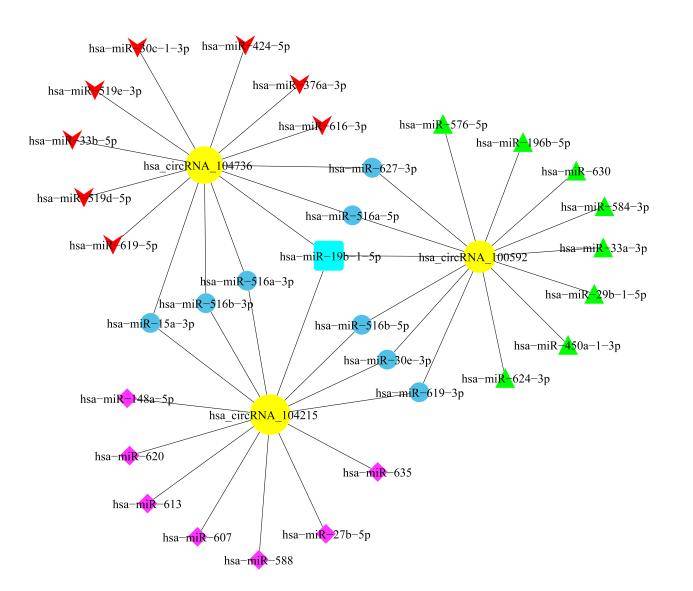
Construction of circRNA/LncRNA-miRNA-mRNA Network
Certain circRNA/LncRNA molecules can regulate the expression of miRNA target genes by binding to miRNAs. Through the analysis of circRNA/LncRNA-miRNA-mRNA associations, we can assist in deducing the circRNA/LncRNA molecules acting as miRNA sponges and their mechanisms of action.
Our unique combination of long and short reads, single and paired-end sequencing, strand specificity, and capacity for tens of millions to billions of reads per run allow us to sequence your samples in diverse ways. Our experienced personnel can help you to define how our services can be best leveraged for your project, and our strict quality control can ensure you the integrity of delivered results. Please feel free to contact our specialists and discuss suitable strategies for library preparation and sequencing.
Circular DNA tumor viruses make circular RNAs
Proceedings of the National Academy of Sciences | 2018Repeated immunization with ATRA-containing liposomal adjuvant transdifferentiates Th17 cells to a Tr1-like phenotype
Journal of Autoimmunity | 2024Role of the histone variant H2A.Z.1 in memory, transcription, and alternative splicing is mediated by lysine modification
Neuropsychopharmacology | 2024FAK loss reduces BRAFV600E-induced ERK phosphorylation to promote intestinal stemness and cecal tumor formation
Elife | 2023

