
 Sample Submission Guidelines
Sample Submission Guidelines
oxBS-seq
DNA methylation and hydroxymethylation are important epigenetic modifications for gene expression regulation. DNA methylation typically represses gene transcription. Hydroxymethylation, on the contrary, activates gene expression or prompts DNA demethylation.
5-Hydroxymethylcytosine (5hmC) is converted from 5-methylcytosine (5mC) by a group of enzymes termed ten-eleven translocation (TET) family dioxygenases. The gold-standard bisulfite conversion technologies to study DNA methylation do not distinguish between 5mC and 5hmC. However, new approaches to mapping 5hmC genomewide have advanced rapidly, although it is unclear how the different methods compare in accurately calling 5hmC.
CD Genomics can provide three 5hmC genome-wide detection approaches:
- Whole-genome bisulfite/oxidative bisulfite sequencing (WGBS/WGoxBS-seq)
- Reduced representative bisulfite oxidative bisulfite sequencing (RRBS/RRoxBS)
- Antibody-based immunoprecipitation and sequencing of hydroxymethylated DNA (hMeDIP-seq).
- Targeted DNA bisulfite/hydroxymethylation sequencing (TBS/ oxTBS-seq)
- 5hmC selective chemical labelling technology (5hmC -Seal)
The Introduction of oxBS-seq
5-methyl cytosine (5mC) can be oxidised by a group of oxigenases called ten-eleven translocation enzymes (Tets) . Under consumption of oxygen and 2-oxoglutarate, these Fe(II) dependent dioxigenases oxidise 5mC in a first reaction to 5-hydroxymethyl cytosine (5hmC), followed by 5-formyl cytosine (5fC) and finally 5-carboxy cytosine (5caC). The most abundant formof these oxidised cytosine variants is 5hmC.
Oxidative bisulfite conversion (oxBS), a pre-bisulfite oxidation reaction converts 5hmC to 5fC, which will be converted by bisulfite to 5f-uracil and to thymine in the subsequent PCR. This library is then compared to a traditional bisulfite sequencing library (BS-seq) constructed on the same sample to determine the amount of 5mC and 5hmC for each modified cytosine within the DNA. oxBS-seq has a relatively simple and fast experimental workflow and can obtain both the methylome and hydroxymethylome simultaneously.
If you want to learn more about the introduction of oxBS-seq, you can check out our article: "oxBS-Seq, An Epigenetic Sequencing Method for Distinguishing 5mC and 5hmC".
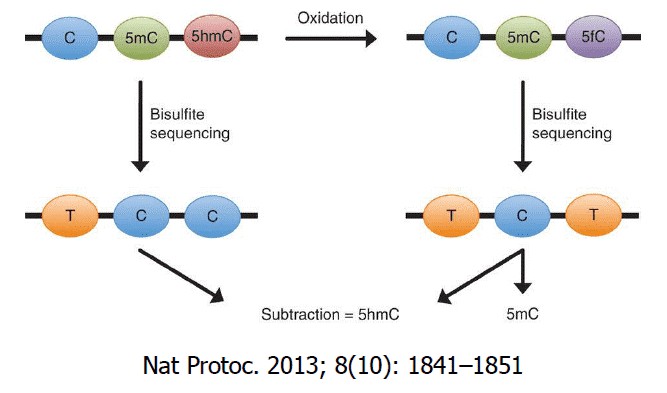 Figure 1. Schematic Diagram of the oxBS-seq Principle. (Booth et al., 2013)
Figure 1. Schematic Diagram of the oxBS-seq Principle. (Booth et al., 2013)
What are the Advantages of oxBS-seq
- Single-nucleotide resolution encompasses both CpG and non-CpG methylation across the entire genome.
- The density of 5mC in repetitive regions and regions of lower density is covered.
- The methods clearly distinguish between 5mC and 5hmC, enabling precise identification of 5mC.
- Establishing a Novel "Gold Standard" for DNA Methylation Detection
- Genome-wide single-nucleotide detection of DNA hydroxymethylation modifications
- Multi-criteria validation for high oxidation efficiency and high bisulfite conversion rate
- Experimental bias is minimal, with high reproducibility (R²>0.98)
- Satisfying diverse sequencing application needs: Streamlined genome-wide oxidation methylation sequencing (oxRRBS), Targeted region oxidation methylation sequencing (Target-oxBS)
What are the Application of oxBS-seq
Our oxBS-seq analysis presents versatile applications across various research domains:
- Epigenetics: Facilitating investigations into the interplay between DNA methylation and hydroxymethylation across diverse biological scenarios, our oxBS-seq analysis offers a nuanced understanding of epigenetic modifications intertwined with gene regulation, cellular differentiation, and developmental pathways.
- Disease Research: Through meticulous examination of DNA methylation and hydroxymethylation patterns within disease samples, our oxBS-seq analysis unveils epigenetic alterations linked with afflictions including cancer, neurological disorders, and cardiovascular ailments.
- Environmental Epigenomics: Widely employed to scrutinize the influence of environmental stimuli on DNA methylation and hydroxymethylation dynamics, our oxBS-seq analysis aids in discerning epigenetic shifts induced by varying environmental exposures, thereby elucidating their potential implications on health and disease susceptibility.
- Developmental Biology: Facilitating the exploration of DNA methylation dynamics throughout embryonic development and tissue differentiation processes, our oxBS-seq analysis furnishes intricate methylation landscapes at high resolutions.
oxBS-seq Sequencing Workflow
Genomic DNA was processed to achieve 10 kb fragments. Fragmented DNA was concentrated using purification columns. This fragmented and purified DNA was taken forward for oxidation + bisulfite treatment. Buffer exchange, denaturation, oxidation, bisulfite conversion, cleanup, and qualitative assessment of 5-hydroxymethylcytosine oxidation was carried out. Last, oxBS- and BS-converted DNA were used to construct oxBS-seq and corresponding BS-seq libraries. All double and single stranded DNA was quantified on an Agilent 2100 Bioanalyser before cluster generation and sequencing on illumina platform according to the manufacturer's protocols.
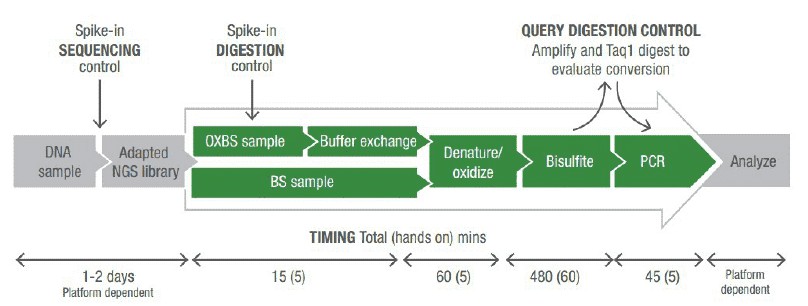 Figure 2. Schematics of steps for high-throughput sequencing of the Ig sequence repertoire (Georgiou et al., 2014).
Figure 2. Schematics of steps for high-throughput sequencing of the Ig sequence repertoire (Georgiou et al., 2014).
Service Specification
Sample Requirements
|
|
 |
Sequencing
|
 |
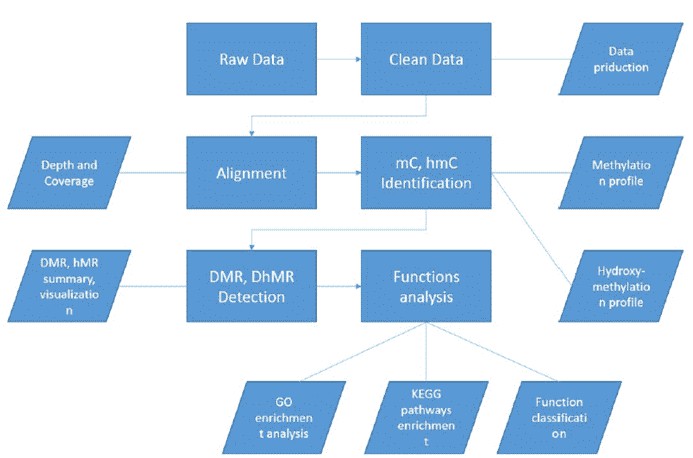
Bioinformatics Analysis We provide customized bioinformatics analysis including:
|
Analysis Pipeline
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in oxBS-seq for your writing (customization)
References:
- Giehr, Pascal, Kyriakopoulos, et al. Two are better than one: HPoxBS - hairpin oxidative bisulfite sequencing. Nucleic acids research, 2018.
- Skvortsova K, Zotenko E, Luu P L, et al. Comprehensive evaluation of genome-wide 5-hydroxymethylcytosine profiling approaches in human DNA. Epigenetics & Chromatin, 2017, 10(1):16.
- Booth M J, Ost T W B, Beraldi D, et al. Oxidative bisulfite sequencing of 5-methylcytosine and 5-hydroxymethylcytosine. Nature protocols, 2013, 8(10): 1841-1851.
- Han Y, Ji L, Guan Y, et al. An epigenomic landscape of cervical intraepithelial neoplasia and cervical cancer using single‐base resolution methylome and hydroxymethylome. Clinical and translational medicine, 2021, 11(7): e498.
Demo Results
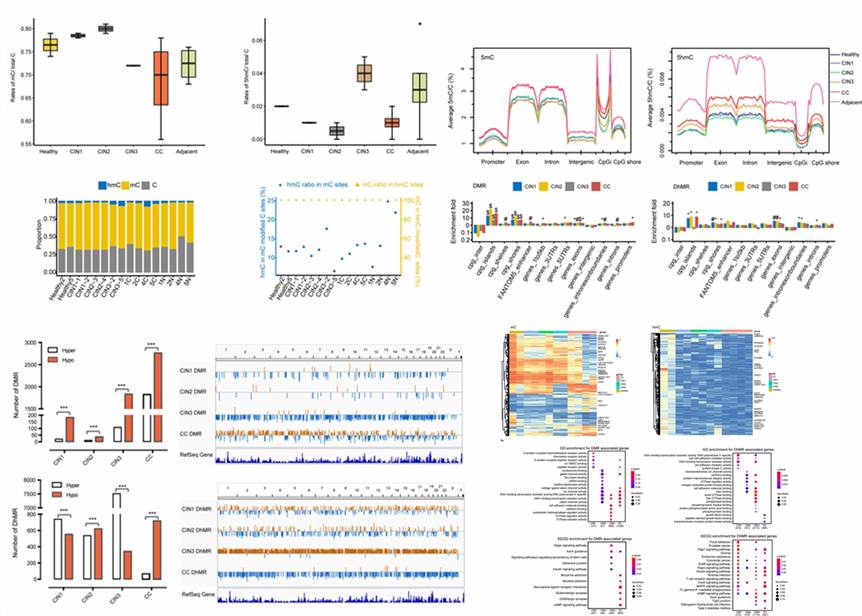 (Han et al., 2021)
(Han et al., 2021)
oxBS-seq FAQs
1. What is oxBS-seq?
oxBS-seq is a high-throughput sequencing technique used to measure the distribution of DNA methylation and hydroxymethylation. It combines oxidative bisulfite treatment (oxBS) with sequencing analysis to distinguish 5mC and 5hmC at single-base resolution, providing detailed information about these two modifications in the genome.
2. How does oxBS-seq differ from traditional BS-seq?
Traditional BS-seq (Bisulfite sequencing) converts unmethylated cytosines (Cs) to uracils (Us) while preserving methylated Cs. However, 5hmC is also converted to T by BS, making it indistinguishable from 5mC. In contrast, oxBS-seq chemically oxidizes 5hmC to C before BS treatment, allowing differentiation between 5mC and 5hmC.
3. What is the workflow of oxBS-seq?
The oxBS-seq workflow encompasses several key steps: DNA extraction, oxidative methylation treatment, bisulfite (BS) treatment, sequencing, and subsequent data analysis. Initially, 5hmC undergoes oxidation to C, followed by BS treatment to convert unmethylated Cs to Ts while preserving 5mC and 5hmC. Subsequently, sequencing data is generated and subjected to comprehensive analysis utilizing bioinformatics tools.
4. What are the analysis methods for oxBS-seq data?
The analytical rubrics applied to oxBS-seq data entail several aspects: preprocessing, alignment, appraisement of methylation levels and differential modification analysis. The step of preprocessing extends to the tasks of quality control and sequence trimming. Alignment is a task that facilitates matching the sequencing data to a reference genome, enabling the estimation of methylation levels at each site and permitting differentiation between 5mC and 5hmC. An array of subsequent statistical analyses and biological interpretations are deployed to bring to light conclusions germane to the apical research question.
5. How to design oxBS-seq experiments?
Strategizing oxBS-seq experiments demands the execution of certain tasks such as sample selection, devising the experimental design, determining the sequencing depth, and strategizing data analysis. To holistically address an array of research queries, the experimenter may fine-tune the experiment parameters, thereby ensuring enhanced experimental result reliability and accuracy. Additionally, it is essential that efficacious experimental controls and replicates are incorporated, serving to endorse reproducibility and stability of findings.
6. What are the limitations of oxBS-seq?
Although oxBS-seq boasts of abundant advantages, it is not without its limitations. For instance, a spectrum of low methylation levels or areas of low genomic complexity may potentially compromise the accuracy of the technique. Furthermore, the technical rigor demanded by the experimental procedures and subsequent data analysis can bring forth formidable challenges.
oxBS-seq Case Studies
Genome-Wide DNA Methylation and Hydroxymethylation Changes Revealed Epigenetic Regulation of Neuromodulation and Myelination in Yak Hypothalamus
Journal: Frontiers in Genetics
Impact factor: 4.772
Published: 27 September 2021
Background
5-Methylcytosine (5mC) and 5-Hydroxymethylcytosine (5hmC) are significant epigenetic modifications integral to the process of neurodevelopment. However, scant research exists that identifies genome-wide patterns of 5mC and 5hmC in animal brain regions as a function of natural high-altitude environments.
Methods
- yak and cattle
- RNA and DNA extraction
- Reduced Representation Bisulfite Sequencing
- Oxidative Reduced Representation Bisulfite Sequencing
- Identification of Differentially Methylated Regions
- Identification of Differentially Hydroxymethylated Regions
Results
RRBS and oxRRBS provide single-base resolution profiling of methylation and hydroxymethylation across 3.28M CpG sites, with oxRRBS showing higher correlation between opposite strands. The lower consistency in RRBS may be due to dynamic 5hmC involvement in DNA demethylation. Analysis focused on "true" 5hmC sites revealed tissue-specific differences between yak and cattle, particularly in the hypothalamus, cerebellum, and brainstem.
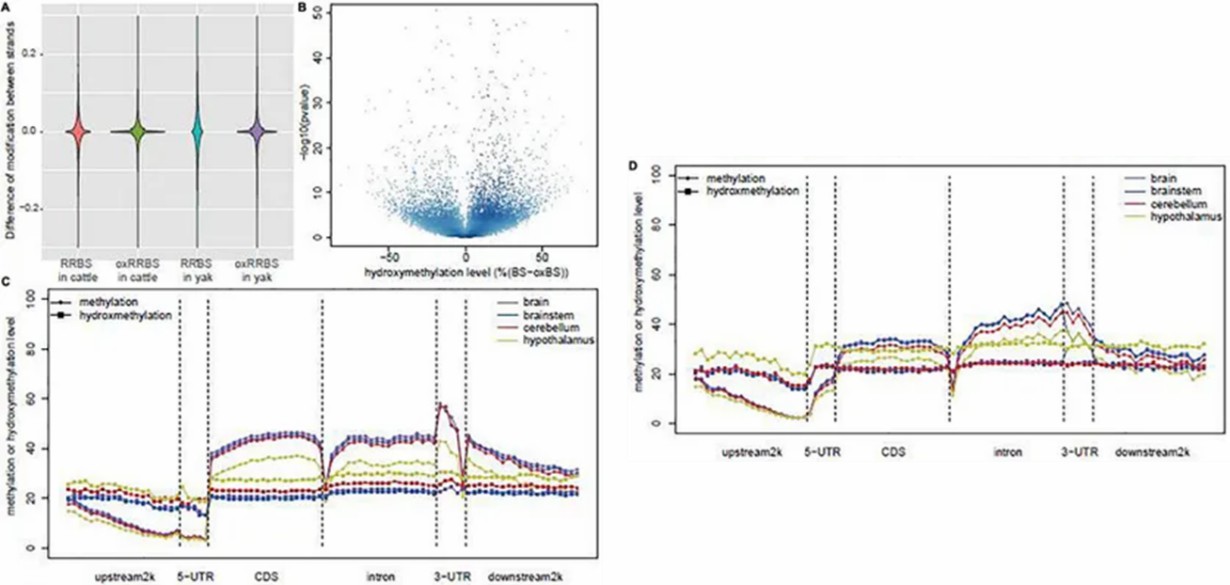 Fig 1. Global DNA methylation and hydroxymethylation among the samples.
Fig 1. Global DNA methylation and hydroxymethylation among the samples.
The study elucidated distinct distribution patterns of 5mC and 5hmC levels across genes, highlighting significant differences between tissues, particularly in the hypothalamus. Pairwise comparisons between yak and cattle brains revealed differential methylation and hydroxymethylation regions (DMRs and DhMRs), mainly driven by altered 5mC and 5hmC levels. Enrichment analysis of gene ontology (GO) terms underscored tissue-specific regulatory mechanisms, particularly in nervous system-related pathways, suggesting intricate epigenetic regulation in brain function across species.
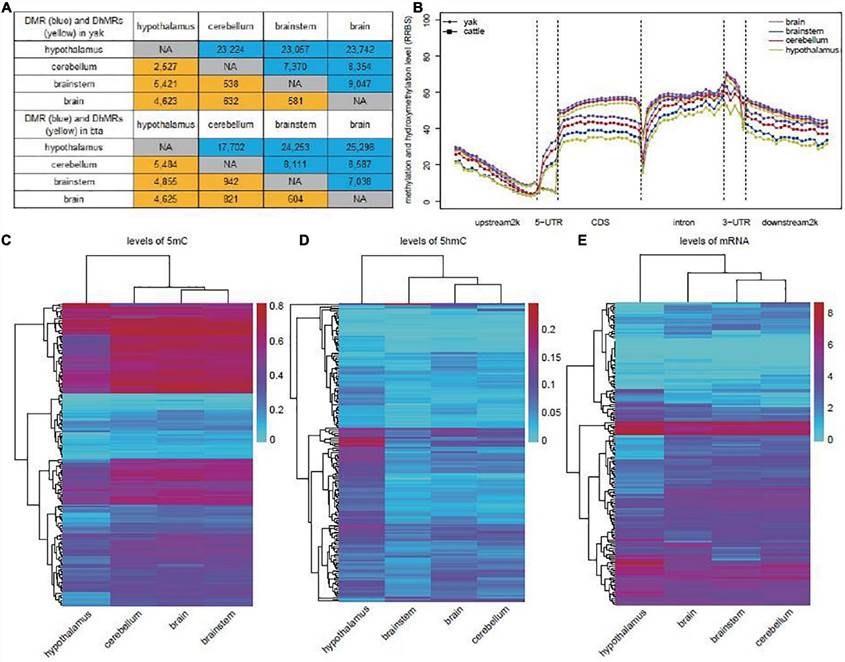 Fig 2. Identification of DMRs and DhMRs in yak and cattle.
Fig 2. Identification of DMRs and DhMRs in yak and cattle.
Conclusion
This study utilizes RRBS and oxidative RRBS (oxRRBS) techniques to uncover significant variances in 5mC and 5hmC present in the hypothalamus and other brain regions of yaks and cows. This includes the identification of differentially methylated regions (DMRs) and differentially hydroxymethylated regions (DhMRs), with most of them overlapping. Consequently, differentially expressed genes (DEGs) potentially regulated by DMRs and DhMRs that may play crucial roles in neural regulation and myelin formation were validated. In summary, the results indicate that the epigenetic regulation mediated by 5mC and 5hmC may extensively impact the development and biological functions of the hypothalamus, possibly contributing to the enhancement of physiological adaptability under high-altitude conditions.
Reference:
- Chai Z, Wu Z, Ji Q, et al. Genome-wide DNA methylation and hydroxymethylation changes revealed epigenetic regulation of neuromodulation and myelination in yak hypothalamus. Frontiers in Genetics, 2021, 12: 592135.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Restriction endonuclease cleavage of phage DNA enables resuscitation from Cas13-induced bacterial dormancy
Journal: Nature microbiology
Year: 2023
IL-4 drives exhaustion of CD8+ CART cells
Journal: Nature Communications
Year: 2024
High-Fat Diets Fed during Pregnancy Cause Changes to Pancreatic Tissue DNA Methylation and Protein Expression in the Offspring: A Multi-Omics Approach
Journal: International Journal of Molecular Sciences
Year: 2024
KMT2A associates with PHF5A-PHF14-HMG20A-RAI1 subcomplex in pancreatic cancer stem cells and epigenetically regulates their characteristics
Journal: Nature communications
Year: 2023
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch
Journal: Science Advances
Year: 2024
Genomic imprinting-like monoallelic paternal expression determines sex of channel catfish
Journal: Science Advances
Year: 2022
See more articles published by our clients.


