
 Sample Submission Guidelines
Sample Submission Guidelines
Introduction to Nanopore Targeted Sequencing
Nanopore Targeted Sequencing enables researchers to focus sequencing effort on the most relevant parts of the genome—whether specific genes, regions with high biological significance, or rare microbial taxa—while avoiding unnecessary sequencing of background DNA.
Unlike traditional methods that require PCR amplification or hybridization capture, nanopore technology offers flexible strategies to perform targeted sequencing:
Nanopore Amplicon-based approaches for cost-effective profiling of PCR products
Cas9-targeted sequencing for precise enrichment of disease- or AMR-related genes
Adaptive sampling, a unique software-driven method, which enriches or depletes DNA fragments during sequencing without any additional lab steps
This flexibility makes nanopore targeted sequencing particularly powerful for applications in oncology, microbiology, epigenetics, and evolutionary studies.
Why Choose Nanopore Targeted Sequencing?
Unbiased, Long-Read Sequencing
Nanopore sequencing supports any read length from 50 bp to >4 Mb, providing the ability to span large structural variants, repetitive elements, and GC-rich regions that short-read methods fail to resolve.
Preserve Epigenetic Information
Unlike PCR- or probe-based enrichment, nanopore targeted sequencing directly analyzes native DNA and RNA molecules, retaining methylation and base modification signals essential for regulatory and cancer research.
Real-Time Enrichment
Adaptive sampling allows researchers to monitor data output during the run, enriching or depleting reads of interest in real time. This reduces turnaround time and ensures efficient use of sequencing capacity.
Flexible, Cost-Effective Workflows
With no need for expensive capture panels or extensive optimization, nanopore targeted sequencing provides a faster and more scalable path to results compared to legacy methods.
| Feature | Nanopore Targeted Capture | Nanopore Whole Genome | Illumina Targeted Capture |
| Read Length | Long reads | Long reads | Short reads |
| Variation | Complex structural variations epigenetic modifications |
whole-genome variations epigenetic modifications |
SNVs/Indels |
| Real-time Sequencing | Yes | Yes | No |
| Amplification Bias | No | No | Yes |
| Portability | High | High | Low |
| Throughput | Moderate | Moderate | High |
| Cost | Moderate | High | Moderate |
Our Nanopore Targeted Sequencing Service Packages
At CD Genomics, we offer carefully designed service packages for Nanopore Targeted Sequencing to meet diverse research requirements. Each package falls under the umbrella of targeted sequencing — selecting specific genomic regions of interest — but differs in methods, input requirements, and delivered data. Review the options below and select the package that aligns best with your project goals, sample quality, and budget.
| Package Name | Best For / Use Cases | Input & Sample Requirements | What's Included | Advantages |
|---|---|---|---|---|
| Basic Amplicon Package | Projects focused on specific hotspot mutations, small gene panels, 16S / ITS barcoding, microbial marker identification | DNA ≥ 50–100 ng; acceptable fragment size ≥ ~500-1000 bp; targets well known; moderate number of loci | PCR primer design; PCR amplification; nanopore sequencing of amplicons; SNV / small indel calling; basic QC report | Low cost; fast turnaround; well-suited for labs with limited sample material; high depth over few targets |
| Cas9 / Panel Deep-Target Package | Medium to large gene panels; structural variant detection; challenging or repetitive regions; preservation of methylation and epigenetic features | High quality DNA (≥ 200-500 ng), long fragment size preferred (>10-20 kb); minimal degradation; sufficient sample purity | Guide RNA design; Cas9-mediated enrichment; long-read nanopore sequencing; SNV + SV calling; optional methylation profiling; detailed report | Probe‐free targeting of difficult regions; retains native DNA modifications; capable of detecting large insertions / deletions and mobile elements |
| Adaptive Sampling Package | Projects needing flexibility: changing target coordinates on the fly; host / background DNA depletion; rare targets in mixed samples | Genomic DNA of good quality; quantity varies by target size; input requirement depends on proportion of genome targeted; BED file of target coordinates provided | Setup of adaptive sampling in MinKNOW; sequencing with on-device enrichment or depletion; real-time monitoring; SNV, SV, base modification analysis; QC & coverage statistics | No extra wet lab enrichment; rapid setup; dynamic targeting; good for environmental or metagenomic samples with background DNA issues |
| Comprehensive Panel + Multi-Omics Package | Testing many genes (e.g. hereditary cancer panels), integrating mutation, structure, methylation, and haplotype data; large studies requiring full feature set | High input (≥ ~500 ng–1 µg depending on targets); high DNA integrity; long fragments strongly desirable; sample QC required | Full panel enrichment (via Cas9 or hybrid probe designs); long-read data; SNV / SV / indel calling; DNA methylation / epigenetic modifications; haplotype phasing; full QC / annotation; comprehensive reporting | Rich data in one experiment; covers all variant types + epigenetic marks; useful for in-depth disease genetics, trait mapping, functional genomics |
Which Package Should You Choose?
- If your targets are few, well characterized, and you need fast results → Basic Amplicon Package will often suffice.
- If structural variants, repeats, or difficult genomic regions matter, choose the Cas9 / Panel Deep-Target or Comprehensive Panel + Multi-Omics package.
- If sample is mixed / background DNA high / you expect to adjust targets → Adaptive Sampling gives more flexibility.
- For projects requiring all layers of insight (mutations + structure + methylation + haplotypes), the Comprehensive Package offers the fullest data set.
Applications of Nanopore Targeted Sequencing
Cancer Genomics
- Detect structural variants such as large insertions, deletions, and inversions with breakpoint resolution
- Profile epigenetic modifications (e.g., MLH1 promoter hypermethylation in Lynch syndrome research)
- Support hereditary cancer risk panels with 250+ genes, including BRCA1/2
- Enable haplotype phasing for allele-specific risk assessment
Antimicrobial Resistance (AMR)
- Track ARGs such as blaCTX-M and blaTEM with Context-seq
- Place resistance genes in their genomic context (plasmids, transposons)
- Study cross-species transmission of ARGs between humans, animals, and environment
Microbiome and Metagenomics
- Remove host DNA to enrich microbial genomes
- Enrich rare taxa or functional genes (nifH, amoA, ARGs) for ecological studies
- Assemble near-complete microbial genomes from low-abundance species
Epigenetics and Gene Regulation
- Target promoters, enhancers, or imprinting regions for direct methylation profiling
- Study repeat elements such as LINE-1 to understand genome stability
- Map allele-specific methylation patterns in cancer or developmental biology
Ancient DNA and Forensics
- Maximize use of degraded or limited samples
- Enrich mitochondrial genomes, sex-determination loci, or species markers
Synthetic Biology
- Verify artificial chromosomes, engineered pathways, or CRISPR edits
- Target large constructs to confirm integrity and insertion points
Our Nanopore Targeted Sequencing Workflow
Sample submission & quality control
- Accept blood, tissue, cells, microbial samples, and ancient DNA.
- DNA QC: purity, integrity, and concentration checks.
Library preparation
- Amplicon, Cas9-based, or standard ligation protocols.
Nanopore sequencing run
- Scalable throughput on GridION or PromethION.
Real-time monitoring & adaptive sampling
- On-device selection for targets or host depletion.
Bioinformatics analysis
- SNV/Indel calling, SV detection, methylation analysis, haplotype phasing.
Report delivery
- Comprehensive dataset, annotated variants, and publication-ready results.
 Workflow of CD Genomics Nanopore Targeted Sequencing, from DNA extraction and target definition to enrichment, long-read sequencing, and bioinformatics reporting.
Workflow of CD Genomics Nanopore Targeted Sequencing, from DNA extraction and target definition to enrichment, long-read sequencing, and bioinformatics reporting.
Nanopore Targeted Sequencing Bioinformatics Analysis
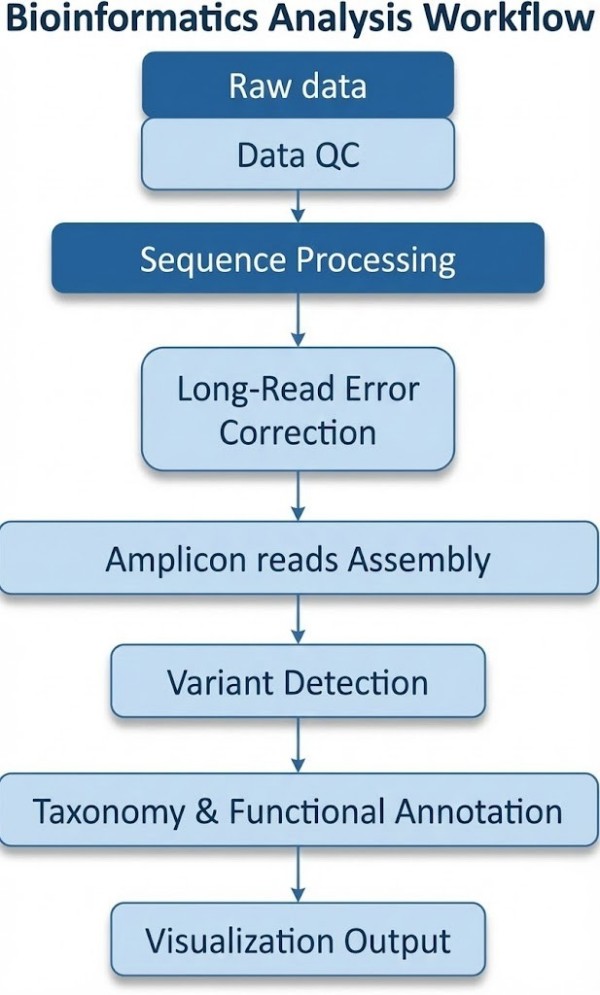
Deliverables
- Raw sequencing files (FASTQ)
- Alignment (BAM) and variant calls (VCF)
- Coverage/enrichment statistics
- Annotated reports with SNVs, SVs, methylation (PDF + Excel)
- Graphical summaries (circos plots, variant maps, methylation tracks)
Why Partner with CD Genomics?
Years of expertise in genomics sequencing across Illumina, PacBio, and Nanopore platforms
CRO-grade reliability, trusted by pharma, biotech, and academic institutions
Flexible project design: from single targets to full cancer gene panels
Comprehensive bioinformatics support: variant annotation, methylation mapping, AMR gene tracking
End-to-end service: sample QC, sequencing, data analysis, secure data portal delivery
Sample Requirements
Acceptable Sample Types & Inputs
| Sample Type | Recommended Quantity/Input | Minimum Quantity/Input | Minimum Concentration / Purity | Notes on Integrity / Handling |
|---|---|---|---|---|
| High-Molecular-Weight Genomic DNA | ≥ 5 µg in total, concentration ≥ 20-50 ng/µL | ≥ 3 µg | OD 260/280 ≈ 1.8; OD 260/230 ≈ 2.0-2.2; RNase-treated; free of visible protein, phenol, detergent contamination | Average fragment size large (ideally ≥ 30 kb); minimal degradation; stored in a buffer such as 10 mM Tris pH 8.0-8.4; shipped cold (ice-packs or dry-ice) |
| Microbial / Environmental DNA | ≥ 500 ng | ≥ 300-500 ng | ≥ ~10 ng/µL; high purity (no inhibitors) | If sample includes host or contaminants, consider host depletion or choosing adaptive sampling paths; properly frozen or stabilized during transport. |
| Tissue or Cell Samples | Enough input to yield high-molecular-weight DNA (typically ≥ few µg) | As high as possible; degraded or low-yield samples may compromise long reads | Purity same as above; DNA free of RNA / protein contaminants; minimal DNA degradation | Fresh or snap frozen preferred; avoid repeated freeze-thaw; for tissues, thinner slices help; storage and transport on dry ice or blue ice. |
Buffer, Storage & Shipping Conditions
- DNA may be in RNase-free water, TE buffer, or 10 mM Tris (pH 8.0-8.4). Avoid buffers with detergents, surfactants, or high EDTA that may interfere with downstream steps.
- For DNA in aqueous buffer or Tris/TE, ship with ice packs or dry ice to keep cold. For DNA stored in 70% ethanol, room temperature shipping may be acceptable.
- Label sample tubes clearly: use permanent marker; label both top and side; sample names on tubes must match those on the submission form. Use sturdy tubes (1.5 mL recommended) or plates with sealed lids; protect against breakage.
- Quality Control Metrics
- Purity: OD260/280 close to ~1.8; OD260/230 around 2.0-2.2. DNA integrity: minimal degradation; visual assessment on gel or by pulsed-field or equivalent; high molecular weight (size distribution skewed to long fragments).
- Concentration: measured preferably by fluorescent methods (e.g., Qubit) rather than only by UV absorbance; ensure sufficient input for the chosen package.
Submission Form, Labeling & Documentation
- Complete the Sample Submission Form, listing sample ID, sample type, quantification and QC metrics. The sample ID on the form must exactly match the label on the tube.
- Submit electronic version of QC data (e.g., concentration, OD ratios, fragment size profile) together with physical samples.
Demo Results
Taxonomic Resolution Improvement

Comparison of microbial classification resolution between short-read and nanopore long-read amplicon sequencing.
Consensus Accuracy with Error Suppression
- Consensus polishing improves variant detection accuracy in nanopore targeted sequencing.

Community Diversity Analysis

Adaptive sampling enhances detection of rare microbial taxa by depleting host DNA background.
Structural Variant & Repeat Resolution

Cas9-targeted enrichment reveals structural variants and repeat expansions with breakpoint resolution.
Epigenetic Insights from Native DNA

Direct detection of DNA methylation during nanopore targeted sequencing of native DNA.
Nanopore Target Seq FAQs
Q1: What is nanopore targeted sequencing and how does it differ from whole-genome sequencing?
Nanopore targeted sequencing focuses on specific genomic regions of interest—such as genes, hotspots, or structural variant breakpoints—rather than sequencing the entire genome. This approach enables higher coverage of those targets, less data waste, and retention of long reads and epigenetic modifications. Whole-genome sequencing provides comprehensive coverage but is more costly and generates more data than needed for focused studies.
Q2: What are the different methods or "routes" to do targeted sequencing with nanopore technology?
There are several approaches: (1) PCR-based amplicon sequencing for well-defined small targets; (2) probe or hybrid capture panels (less common in nanopore but available via custom or third-party designs); (3) CRISPR-Cas9 enrichment, which is amplification-free and suited for structural variants and difficult genomic regions; and (4) adaptive sampling, which enriches or depletes reads in real time using software without additional wet-lab steps.
Q3: What is the difference between nanopore amplicon sequencing and other targeted sequencing methods?
Amplicon sequencing is PCR-based, cost-effective, and rapid, but it may introduce amplification bias, miss GC-rich or repetitive regions, and remove methylation information. Cas9 enrichment or adaptive sampling avoids PCR, preserves methylation, and captures structural complexity across long genomic regions.
Q4: How does nanopore Cas9 targeted sequencing compare with hybrid capture?
Cas9 enrichment is faster, simpler, and probe-free, producing long reads that resolve repeats and structural variants better. Hybrid capture is useful for broad panels, but requires probe design, more steps, and may not capture complex structural contexts as effectively.
Q5: Can nanopore targeted sequencing detect epigenetic modifications such as DNA methylation?
Yes. When using amplification-free methods like Cas9 enrichment or adaptive sampling, nanopore sequencing directly detects base modifications, including methylation, without additional chemical treatments or library preparation.
Q6: Can adaptive sampling be applied to low-input or degraded DNA?
Yes, adaptive sampling works with standard nanopore libraries. For highly fragmented or low-input DNA, such as FFPE or ancient samples, modified protocols can help improve performance, although enrichment efficiency depends on fragment size and quality.
Q7: What are the limitations or trade-offs of nanopore targeted sequencing compared to short-read or PCR-based methods?
Nanopore sequencing may have lower single-read accuracy than short-read platforms, though coverage and consensus improve results. Amplicon methods can suffer from PCR bias, while adaptive sampling efficiency depends on target size and DNA length. In contrast, nanopore sequencing delivers long reads, preserves methylation, and resolves structural variants and repeats that short-read or PCR-only approaches cannot.
Q8: What sample quality and input are required for reliable targeted sequencing results?
High molecular weight DNA is preferred, with minimal degradation, proper purity ratios (OD 260/280 ~1.8; OD 260/230 ~2.0–2.2), and sufficient concentration. Amplicon sequencing tolerates smaller or more fragmented inputs, while Cas9 enrichment and adaptive sampling perform best with longer DNA fragments and high-quality preparations.
Q9: Which nanopore device should I choose for targeted sequencing projects?
Device selection depends on scale and complexity. MinION is suitable for small panels or individual targets, while GridION or PromethION are recommended for larger panels, multiple samples, or projects requiring higher throughput. Multiplexing options, flow cell type, and read length goals also influence device choice.
Q10: How is adaptive sampling set up and what does it do for targeted sequencing?
Adaptive sampling is configured in MinKNOW by providing reference files and optional BED coordinates for target regions. During sequencing, software evaluates read starts in real time, allowing on-target reads to continue while rejecting off-target reads to free pores. This provides enrichment or depletion without extra wet-lab enrichment, with effectiveness depending on target size and input DNA quality.
Q11: How many variants can be detected reliably with nanopore targeted sequencing?
Nanopore targeted sequencing can detect SNVs, small indels, structural variants such as large deletions or inversions, repeat expansions if reads span them, and haplotypes when coverage and read length allow. Sensitivity and specificity depend on target design, sequencing depth, and DNA quality.
Q12: What bioinformatics deliverables are included in a standard project?
Typical deliverables include raw reads (FASTQ), alignments (BAM), variant calls (VCF for SNVs, indels, SVs), coverage and enrichment statistics, optional methylation or base modification files, and graphical summaries such as variant maps or structural variant plots. Annotation reports linking results to genes, known variants, or pathways are also provided.
Nanopore Target Seq Case Studies
Source: Weiwei Gao, Chen Yang, Tianzhen Wang, Yicheng Guo, Yi Zeng (2024). Nanopore-based targeted next-generation sequencing of tissue samples for tuberculosis diagnosis. Frontiers in Microbiology 15:1403619. DOI: 10.3389/fmicb.2024.1403619.
1. Background
Diagnosing tuberculosis (TB) in tissue (extrapulmonary TB, EPTB) is difficult when sputum is unavailable. Traditional methods (HE staining + AFB, HE + PCR, HE + IHC, Xpert MTB/RIF) often yield low sensitivity with tissue samples. This study investigates whether nanopore-based targeted next-generation sequencing (tNGS) can improve diagnostic sensitivity and specificity in tissue samples, including detection of potential drug resistance.
2. Methods
- Total of 110 tissue clinical samples were collected.
- The tNGS assay targeted Mycobacterium tuberculosis. Benchmarked against HE + AFB stain, HE + PCR, HE + immunohistochemistry (IHC with anti-MPT64), and Xpert MTB/RIF.
- Also included drug resistance detection in patients whose samples showed positivity.
3. Results
- tNGS achieved sensitivity 88.2% and specificity 94.1% in tissue samples.
- The comparator methods had notably lower sensitivity: HE + AFB (30.1%), HE + PCR (49.5%), HE + IHC (47.3%), Xpert MTB/RIF (59.1%). Specificities of comparators ranged from ~82-94%.
- Drug resistance analysis: of 93 TB-positive patients, 10 (≈10.75%) had potential drug resistance identified by tNGS.
 Figure 1. Sensitivity and specificity of tNGS versus conventional diagnostic methods for TB in tissue samples (HE + AFB, HE + PCR, HE + IHC, Xpert MTB/RIF.
Figure 1. Sensitivity and specificity of tNGS versus conventional diagnostic methods for TB in tissue samples (HE + AFB, HE + PCR, HE + IHC, Xpert MTB/RIF.
4. Conclusions
Nanopore tNGS on tissue samples performs substantially better than traditional histopathology + molecular methods for diagnosing TB, including in extrapulmonary tissue where bacterial load is low. It also provides drug resistance insights, making it a valuable tool for rapid and accurate diagnosis in clinical settings. The study supports using tNGS as a complement to existing diagnostics for TB.
References:
- Gao W, Yang C, Wang T, Guo Y, Zeng Y. Nanopore-based targeted next-generation sequencing of tissue samples for tuberculosis diagnosis. Front Microbiol. 2024 Jul 3;15:1403619. doi: 10.3389/fmicb.2024.1403619. PMID: 39027106; PMCID: PMC11256091.
- Yang C, Gao W, Guo Y, Zeng Y. Nanopore-based targeted next-generation sequencing (tNGS): A versatile technology specialized in detecting low bacterial load clinical specimens. PLoS One. 2025 May 27;20(5):e0324003. doi: 10.1371/journal.pone.0324003. PMID: 40424288; PMCID: PMC12111578.


