
 Sample Submission Guidelines
Sample Submission Guidelines
Single-Cell DNA Sequencing
CD Genomics provides comprehensive whole genome amplification to discover of DNA mutations in single cells.
The Introduction of Single-Cell DNA Sequencing
Random, low-abundance mutations in the genome of somatic cells are difficult to be qualified and characterized. To circumvent this problem and simultaneously account for the mutational heterogeneity within tissues, sequencing of a representative number of single cells is applied.
Whole genome amplification (WGA) technology has emerged as a tool to perform assessment DNA mutations in single, or a limited number of cells. To enable the discovery of true variation in single cells, the WGA of single-copy genomic DNA starting from a single cell is optimized to obtain sufficient amounts of DNA for sequencing. Faced with the problem of limited or insufficient DNA quantity for various downstream analysis, amplified DNA can be directly used for sequencing.
Various WGA techniques have been developed. We provide services that accurately copy the original source DNA with the lowest false positive rate and validated protocol well-suited for detection of copy number variations and single nucleotide variations across the genome from a single cell after whole genome amplification.
What are the Advantages of Single-Cell DNA Sequencing
- Direct detection of gene-level mutations is of great significance for tumor detection.
- DNA exhibits greater stability compared to RNA, affording sufficient time for sample processing.
- The detection sensitivity for identifying rare mutation-bearing cell clones within tissues can achieve levels as low as 0.1%.
- This approach effectively tackles the challenge posed by mixed samples.
- Furthermore, it offers enhanced spatial resolution.
- Looking ahead, the integration of further omics analyses onto single-cell DNA platforms, including cell surface protein profiling, single-cell methylation analysis, RNA-seq, and BCR/TCR sequencing , holds immense promise.
What are the Application of Single-Cell DNA Sequencing
- To discover of CNVs and single nucleotide variations (SNVs) across genome in single cells or ultra-low input
- To obtain a base-by-base view of an exome at single cell resolution
- Revealing Cellular Heterogeneity
- Tracing Embryonic Development and Cell Differentiation
- Investigating Tumor Development and Therapeutic Resistance
- Exploring Immune System Functionality
- Understanding Neural System Structure and Function
- Analyzing Microbial Community Composition
Single-Cell DNA Sequencing Workflow
The advent of cell sorting/partitioning technologies, such as flow cytometry and microfluidic, has made it possible to capture single cells, and WGA is optimized to providing high yields of whole genomic DNA for sequencing. The general workflow for single-cell DNA sequencing is outlined below.
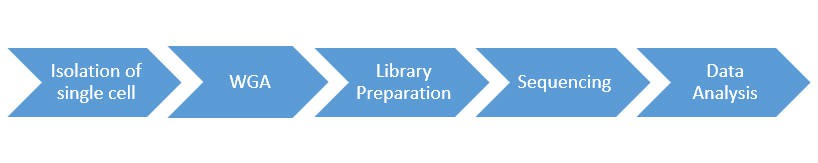
Service Specification
Sample Requirements
|
|
 |
Sequencing
|
 |
Bioinformatics Analysis
|
Analysis Pipeline
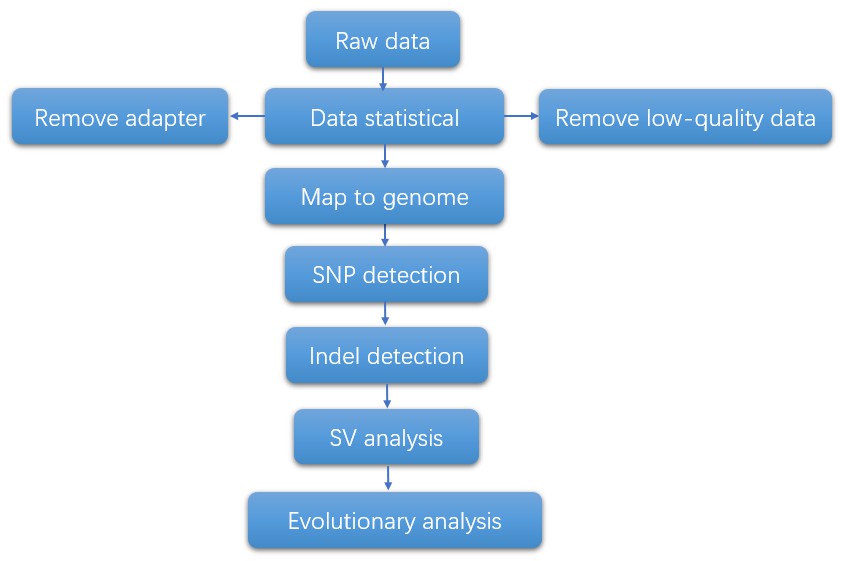
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Single-Cell Sequencing for your writing (customization)
CD Genomics' Single-Cell DNA Sequencing conference focuses on the links between cell variation in tissues and organ function and further elucidates the origins of diseases. If you have additional requirements or questions, please feel free to contact us.
References:
- Deleye, L et al. Performance of four modern whole genome amplification methods for copy number variant detection in single cells. Scientific reports, 2017 Jun 13; 7(1): 3422.
- Dong, X et al. Accurate identification of single nucleotide variants in whole genome amplified single cells. Natare Methods. 2017 May; 14(5): 491–493.
1. What is single-cell DNA sequencing?
Single-cell DNA sequencing refers to a sophisticated DNA sequencing method that involves the analysis of genetic material drawn only from an individual cell. It endows scientists with the capability to perceive and understand the distinctive behavioral patterns exhibited by each cell.
2. Why is single-cell DNA sequencing important?
The importance of single-cell DNA sequencing stems from its intrinsic ability to enable the study and scrutiny of genetic variances present within individual cells coexisting in a single tissue or organism. The awareness of these differences can lay the groundwork in highlighting any irregularities or abnormalities at the cellular level. Consequently, this could pave the way for novel insights into the origin and evolution of diseases, while also assisting in the development of targeted and effective treatment modalities.
3. How is single-cell DNA sequencing different from traditional DNA sequencing methods?
Traditional DNA sequencing methods amalgamate genetic data from a multitude of cells, thereby obscuring the heterogeneity inherent amongst individual cells. In contrast, single-cell DNA sequencing facilitates the examination of singular cells, hence, unearthing this hidden cellular diversity.
4. The principle, advantages and disadvantages of single-cell genome amplification.
i. MDA (Multiple Displacement Amplification)
Invented by the Laskin et al. in 2001. Reacted using random six polymer primers and φ29 DNA polymerase, which had strong chain replacement properties and could amplify the DNA fragment of 50~100kb under isothermal conditions. At the same time, because of its 3 '-5' exonuclease activity and proofreading activity, the φ29 DNA polymerase has high fidelity. The MDA method has a higher genome coverage.
ii. MALBAC (Multiple Annealing and Looping–Based Amplification Cycles)
The Quasilinear amplification process reduces the sequence preference of exponential amplification. The 5 'of amplified primers containing the common sequence of 27bp and 3' is a random sequence of 8bp, which can be combined with the template at low temperature at 15~20 C, and then amplify these ring-shaped amplicons after the quasilinear amplification of 8~12 cycles.
The advantage of MALBAC method is that sequence preference is repeatable between different cells. Because of its better homogeneity of amplification, its data is more suitable for CNV analysis. The weakness of MALBAC is that the fidelity of polymerase it used is not as good as φ29 DNA polymerase, so MALBAC will have more false positives when detecting SNV; in addition, because of its repeatability sequence preference, the region of low amplification in the genome is sometimes lost in the process of amplification.
5. How does single cell DNA sequencing work?
Cell Isolation:
The isolation of individual cells from a heterogeneous population is carried out with precision, employing sophisticated techniques such as fluorescence-activated cell sorting (FACS), microfluidics, or laser capture microdissection.
Genome Amplification:
DNA extracted from the isolated cell undergoes amplification procedures to yield sufficient material for downstream analysis. Established methods like Multiple Displacement Amplification (MDA) and Polymerase Chain Reaction (PCR) are commonly utilized for this purpose.
Library Construction:
Following amplification, DNA molecules are fragmented, and specialized adapters are introduced to facilitate subsequent sequencing steps. This crucial phase involves meticulous processes including DNA shearing, end repair, adapter ligation, and subsequent amplification.
Next-Generation Sequencing (NGS):
The meticulously prepared libraries are then subjected to state-of-the-art next-generation sequencing platforms such as Illumina, PacBio, or Oxford Nanopore. During sequencing, the DNA fragments are meticulously read, yielding comprehensive sequence information.
Data Analysis:
Raw sequencing data undergoes rigorous processing and analysis to identify genetic variants, including single nucleotide variations (SNVs), copy number variations (CNVs), and structural variations (SVs). Advanced bioinformatics tools and algorithms are employed for variant calling, alignment, and meticulous interpretation.
6. What are the techniques for whole genome amplification?
In the realm of whole genome amplification, several prominent technical avenues emerge:
- One such avenue is rooted in PCR methodology, exemplified by DOP-PCR (Degenerate Oligonucleotide-Primed PCR).
- Another path involves isothermal amplification techniques, typified by MDA (Multiple Displacement Amplification).
- Furthermore, methodologies merging PCR with isothermal amplification, such as MALBAC (Multiple Annealing and Looping-Based Amplification Cycles), represent a distinct approach.
- In addition, there exists a category based on transposase mechanisms, exemplified by LIANTI and META-CS.
MDA technology stands out as prevalent and widely adopted within the domain of whole genome amplification. Moreover, MALBAC, harnessing the combined advantages of PCR and isothermal amplification, holds particular significance in fields like single-cell sequencing. While LIANTI and META-CS present promising developments in transposase-based approaches, their widespread adoption and recognition are yet to match that of MDA and MALBAC.
7. What are some challenges of single-cell DNA sequencing?
Inherent challenges consist of the potential loss of genetic material during cell isolation and DNA extraction stages, inaccuracies introduced during the amplification process, and considerable financial investment required for this technology.
8. What is the future outlook for single-cell DNA sequencing?
The future is replete with prospects for single-cell DNA sequencing. Technological advancements promise to decrease expenses and augment throughput, thereby fostering broader usage. The aggregation of single-cell genomic data will also expedite the creation of more sophisticated models of human development and disease progression.
High-throughput, single-microbe genomics with strain resolution, applied to a human gut microbiome
Journal: Science
Impact factor: 60.528
Published: 3 Jun 2022
Background
The human gut microbiome represents a complex and individualized ecosystem characterized by a multitude of microbial species. The presence of diverse strains within a single species can exert significant effects on health, including influencing antibiotic resistance profiles and modulating interactions with the host microbiome. Consequently, a narrow focus on microbial species without consideration of their specific strains may result in the oversight of critical nuances. Despite the importance of strain-level resolution, the genomic composition of the gut microbiome at this level remains largely unexplored, even within an individual.
While shotgun metagenomics offers a comprehensive survey of microbial community genomes, it typically falls short in capturing strain-level variations. Conversely, culture-based methodologies and titer plate-based single-cell sequencing present opportunities to elucidate strain-resolved genomes. However, these approaches are constrained by their limited ability to encompass a broad spectrum of microbial strains.
Methods
- Stool samples
- Microfluidic device fabrication
- Isolation and lysis of microorganisms
- Whole-genome amplification
- Droplet pooling and sequencing library preparation
- Illumina sequencing
- Preprocessing of raw sequencing data
- Genome coassembly
- Phylogeny analysis of genomes
- Diversity of microbiome samples
- Horizontal gene transfer analysis
Results
The researchers employed Microbe-seq to analyze seven gut microbiome samples obtained from a single human subject, resulting in the acquisition of 21,914 single-amplified genomes (SAGs). These SAGs were coassembled into 76 genomes at the species level, with many originating from species that are challenging to culture. Among these genomes, ten species comprised multiple strains, whose genomes were coassembled as well. Utilizing these strain-resolved genomes, the authors reconstructed the horizontal gene transfer (HGT) network within this microbiome. They observed frequent genetic exchange among Bacteroidetes species, particularly associated with a mobile element housing a Type-VI secretion system, known for mediating inter-strain competition.
Furthermore, the droplet-based encapsulation method facilitated the exploration of physical associations between individual microbes and co-localized bacteriophages. Notably, a significant host-phage association was uncovered between crAssphage, the most prevalent bacteriophage documented in the human gut microbiome, and a specific strain of Bacteroides vulgatus.
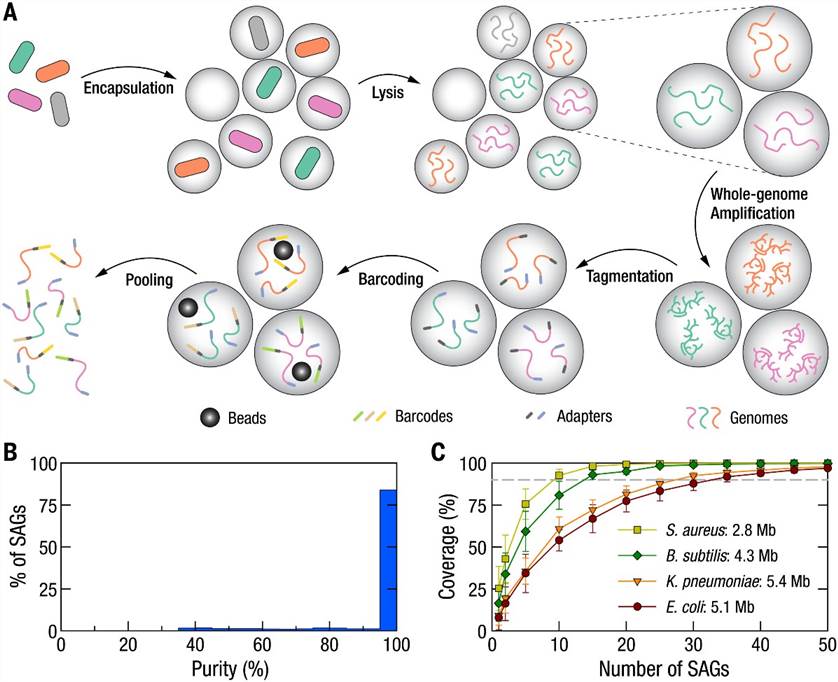 Fig. 1. Schematic of the Microbe-seq workflow and application in a community of known bacterial strains.
Fig. 1. Schematic of the Microbe-seq workflow and application in a community of known bacterial strains.
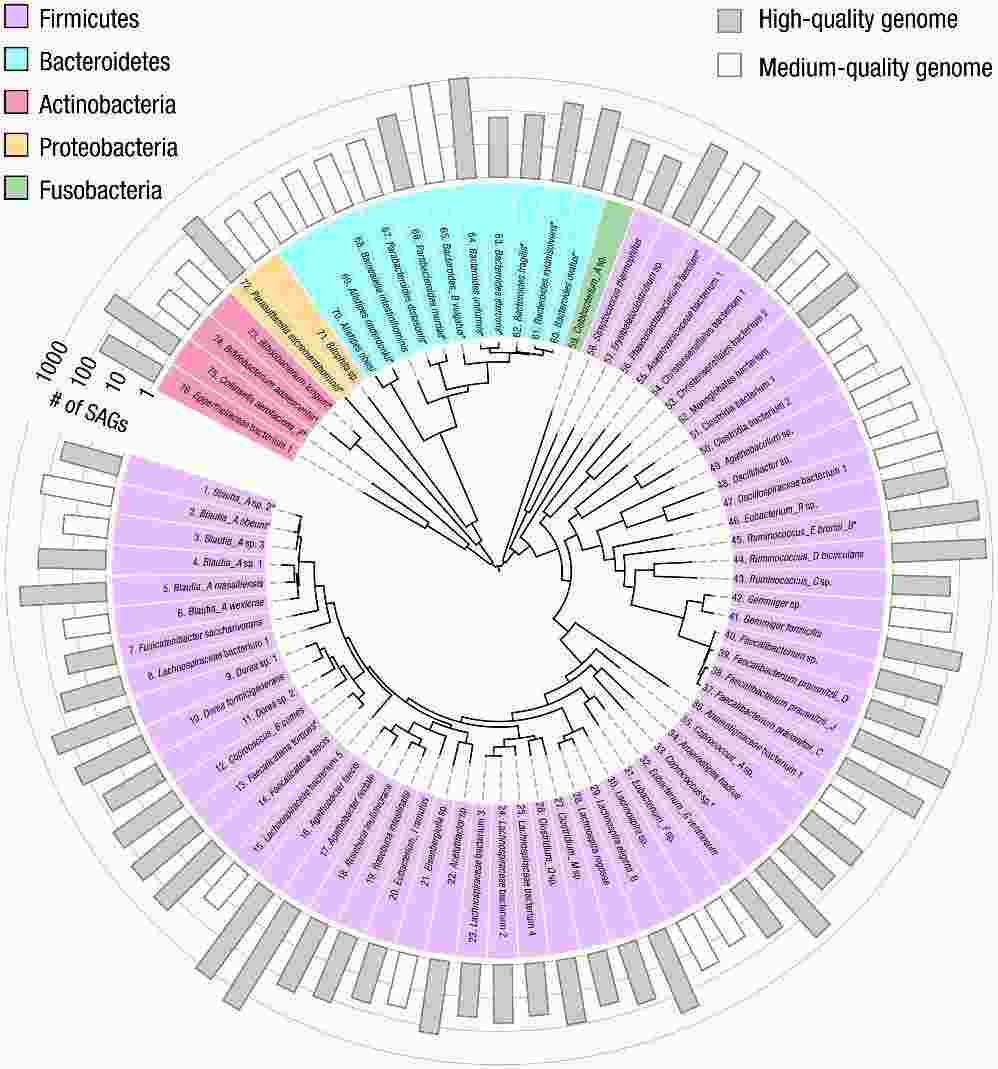 Fig. 2. Coassembled genomes of 76 bacterial species in the human gut microbiome of a single human donor.
Fig. 2. Coassembled genomes of 76 bacterial species in the human gut microbiome of a single human donor.
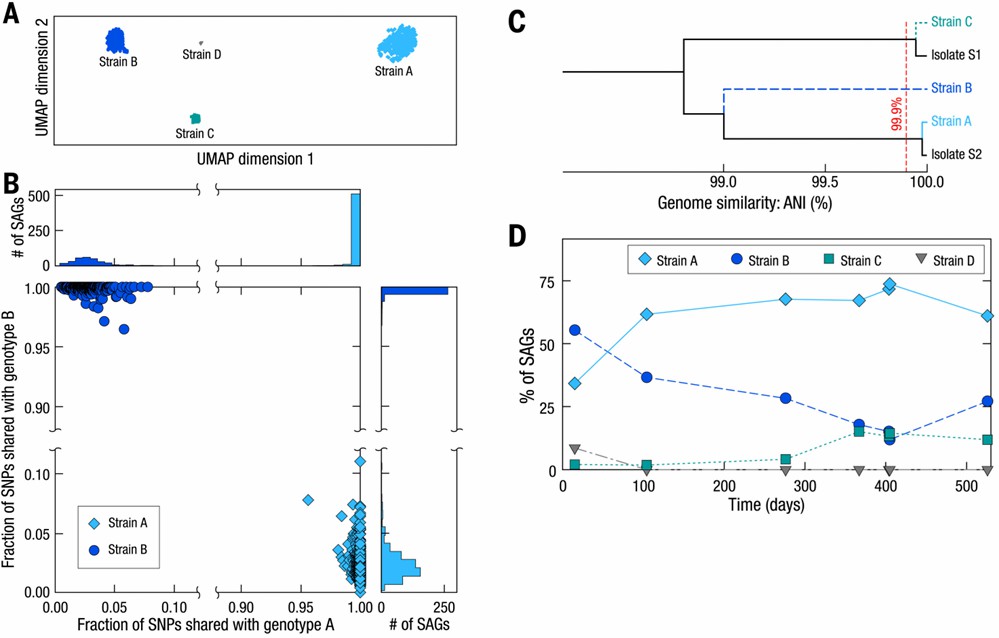 Fig. 3. Strain-resolved genomes of B. vulgatus in the human gut microbiome.
Fig. 3. Strain-resolved genomes of B. vulgatus in the human gut microbiome.
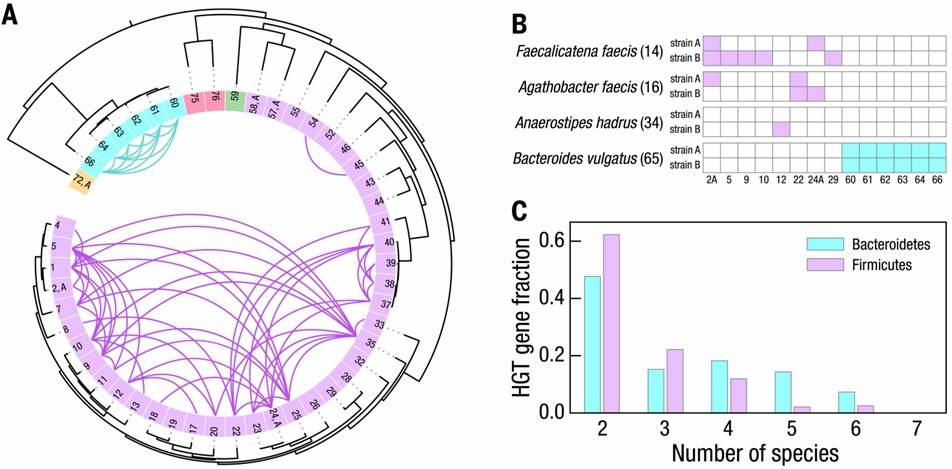 Fig. 4. HGT among bacterial strains within the human gut microbiome of a single donor.
Fig. 4. HGT among bacterial strains within the human gut microbiome of a single donor.
Conclusion
The researchers employed Microbe-seq, a methodology that integrates microfluidic droplet manipulation with customized bioinformatic analysis, to conduct a strain-resolved exploration of the genomic architecture within an individual's gut microbiome. This approach demonstrates versatility and can be readily adapted to investigate diverse microbial ecosystems, including those present in soil and marine environments. Expanding the application of this methodology to a broader human cohort and integrating Microbe-seq with complementary techniques such as functional screening, sorting, and long-read sequencing holds promise for significantly enhancing our understanding of the gut microbiome and its intricate interactions with human health.
Reference:
- Zheng W, Zhao S, Yin Y, et al. High-throughput, single-microbe genomics with strain resolution, applied to a human gut microbiome. Science, 2022, 376(6597): eabm1483.


