
 Sample Submission Guidelines
Sample Submission Guidelines
Viral Metagenomic Sequencing
As an experienced provider of NGS services and a partner of Illumina, CD Genomics is committed to offering unprecedented amounts of sequence data permitting rapid and innovative analytical approaches with cost-effective solutions. We have strong expertise in providing the confident and unbiased viral metagenomic sequencing service by employing state-of-the-art high throughput sequencers, sequencing strategies, and bioinformatics pipelines.
Introduction of Viral Metagenomic Sequencing
Viruses are the most abundant biological entities in the world which outnumber microbial cells 10:1 in most environments. Viruses constantly inhabit our body, and asymptomatic hosts are no exceptions. This emerging vision raises the need for the exploration of the virome. Viral metagenomics can provide insights into the composition and structure of viral communities. Profiling the taxonomic composition of viral communities is important not only for basic research but also for clinical science and practice.
However, viral communities are difficult to be characterized because there is no single gene that is shared by all viral genomes, which limits the application of analogous methods used in bacteria for ribosomal DNA profiling. The low-abundance of free and cellular DNA is another challenge. Metagenomic shotgun sequencing and RNA-seq are two approaches for the characterization of viral communities. Direct sequencing can result in a high background of genetic materials. Therefore, extra procedures are needed to concentrate and purify viral particles (VPs). Multiple viable enrichment methods have been suggested, including filtration, ultracentrifugation, nuclease treatment, multiple displacement amplification (MDA), linker-amplified shotgun library (LASL), and random PCR approach.
Advantages of Viral Metagenomic Sequencing
- No need for cultivation or antibody laboratory tests
- Comprehensive information on community biodiversity, taxonomy, and function
- Promising applications include virus discovery and viral pathogen identification associated with horticulture, veterinary medicine, and human health, especially for difficult-to-diagnose cases.
Application of Viral Metagenomic Sequencing
(1) Disease Treatment: The study of viral diversity, the discovery of unknown viruses, the detection analysis of specific viruses/bacteriophages, and the interaction between bacteria and bacteriophages (in aspects such as the gut, skin, environment, etc. and in relation to health) are vital elements to address. Investigation into the relationships between viruses and diseases (such as metabolic diseases, gastrointestinal diseases, respiratory diseases, allergies, infectious diseases, oral diseases, and tumors) can greatly augment our understanding. Techniques like bacteriophage therapy and sensitization of antibiotic-resistant bacteria could potentially stimulate immune responses.
(2) Food Safety: Monitoring for viruses in food, understanding food-borne viral diversity, investigating pathogens transmitted via food, and monitoring for viruses during food processing are key measures. Tracing viral contamination incidents and studying the link between viruses and the food microbiome can substantially contribute to ensuring food safety.
(3) Livestock Industry: Disease management in animal breeding, such as dealing with diarrhea, swine fever, foot-and-mouth disease, zoonotic diseases, and analyzing feces from wild animals, are significant for maintaining livestock health.
(4) Infectious Disease Monitoring: Monitoring environmental viruses can provide early detection and surveillance clues for disease outbreaks, significantly contributing to public health initiatives.
(5) Environmental Engineering: In environmental engineering pursuits such as wastewater treatment and pollution control, understanding viral communities in water bodies helps evaluate treatment effectiveness and environmental impact. This applies to various environments like public transportation systems, hospitals, oceans, freshwater bodies, air, and glaciers.
(6) Drug Discovery: Researching the virome can reveal viruses that might interact with hosts, providing clues for novel drug discovery.
(7) Agriculture: Studying plant and soil viral diversity could support the development of biopesticides in the agricultural sector.
Viral Metagenomic Sequencing Workflow
Our highly experienced expert team executes quality management by following every procedure to ensure comprehensive and accurate results. The general workflow for viral metagenomic shotgun sequencing is outlined below. Briefly, the basic steps involved in viral metagenomics include the isolation and purification of viral particles by size filtration or density-based centrifugation, sequence-independent amplification of viral nucleic acid, shotgun sequencing or RNA-seq and matched analyses on diversity, taxonomy, and function through a battery of reliable bioinformatics tools.

Service Specifications
Sample requirements
|
|
 |
Sequencing Platforms
|
 |
Bioinformatics AnalysisWe provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
Deliverables
- Quality Control and Host Removal
- Analysis of Reads Species
- Assembly
- Analysis of Assembled Species
- Functional Analysis
- Prediction of Phage Hosts
Apart from the qualified viral metagenomic sequencing, we provide assistance, including the experimental design, the determination of the appropriate sequencing platform, software tools, and analysis methodologies, to accommodate your project. If you have additional requirements or questions, please do not hesitate to contact us, our experienced specialists would like to help you to solve your questions.
References:
- Li Y, et al. VIP: an integrated pipeline for metagenomics of virus identification and discovery. Scientific reports, 2016, 6: 3774
- Rampelli, S. et al. ViromeScan: a new tool for metagenomic viral community profiling. BMC Genomics. 2016, (17):165.
- Lewandowska D W, Zagordi O, Geissberger F D, et al. Optimization and validation of sample preparation for metagenomic sequencing of viruses in clinical samples. Microbiome, 2017, 5(1): 94.
Demo Results
Partial results are shown below:

Per base sequence conten.
Per sequence GC content.

Merged_abundance.

Abundance heatmap of family level.

KEGG_classification.

CAZy function classification.

Boxplot analysis based on bray Curtis (A), binary jaccard (B), unweighted unifrac (C), and weighted unifrac (D).

PCoA analysis based on bray Curtis (A), binary jaccard (B), unweighted unifrac (C), and weighted unifrac (D).

UPGMA clustering tree.
Viral Metagenomic Seq FAQs
1. What are the approaches to whole-genome sequencing of pathogens?
There are currently three main approaches to whole-genome sequencing of pathogens, i.e., metagenomic sequencing, PCR amplicon sequencing and target enrichment sequencing (Figure 1). Direct metagenomic sequencing provides an accurate and integrated representation of the sequences within a sample. PCR amplicon sequencing performs PCR reactions to enrich the viral genome, which substantially increases the workload for large genomes but reduces the costs. Target enrichment sequencing employs virus-specific nucleotide probes bound to a solid phase to enrich the viral genome in a single reaction, which decreases workload but increases the cost compared to PCR amplicon sequencing. The advantages and disadvantages are listed in Table 1. It shows that viral metagenomic sequencing has the unique advantages to investigate pathogen and characterize viral diversity in environmental and clinical samples.
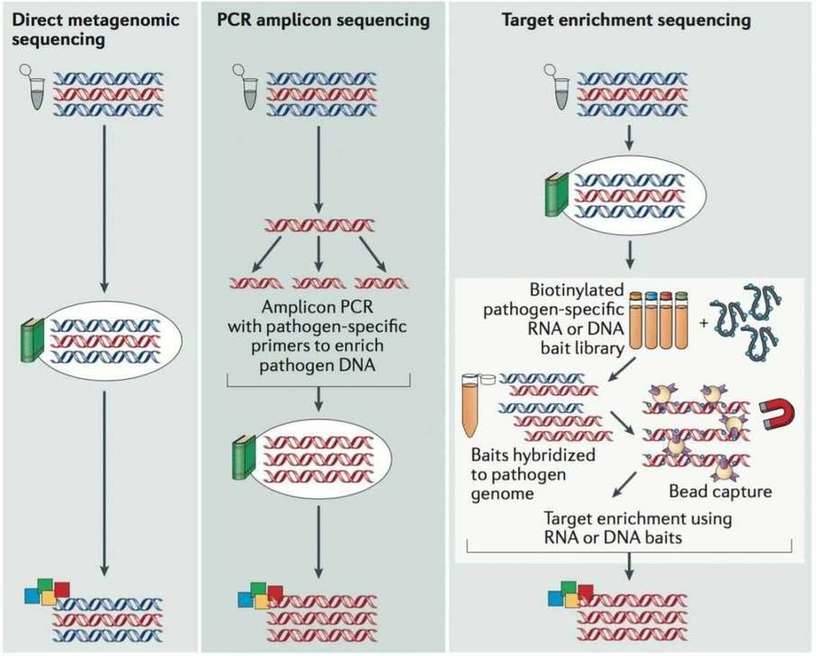 Figure 1. The main approaches used for viral genome sequencing (Houldcroft et al. 2016).
Figure 1. The main approaches used for viral genome sequencing (Houldcroft et al. 2016).
Table 1. Advantages and disadvantages of different viral genome sequencing methods (Houldcroft et al. 2016).
| Method | Advantages | Disadvantages |
|---|---|---|
| Metagenomics |
|
|
| PCR amplicon |
|
|
| Target enrichment |
|
|
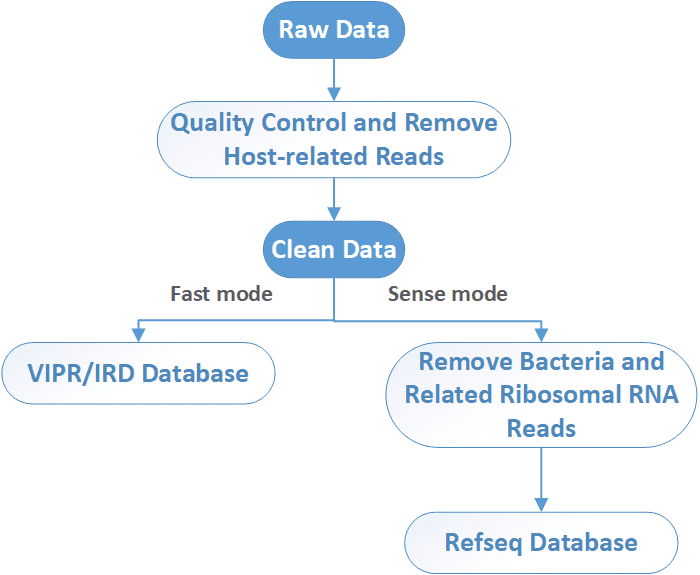
2. What is the general bioinformatics pipeline for viral metagenomic sequencing?
After sequencing, raw NGS reads are first preprocessed by the removal of adapter, low-quality, and low-complexity sequences, followed by computational subtraction of host-related reads. In a fast mode, viruses are identified by alignment to ViPR/IRD nucleotide DB. In a more sense mode, bacteria and related ribosomal RNA reads are removed before alignment to virus database. Unmatched reads are further aligned to a viral protein database (NCBI RefSeq DB). Then the taxonomy identification (Taxl), coverage plot (Covplot), de novo assembly (Multiple k-mer), and phylogenetic analysis (PhyGo) can be performed.
References:
- Houldcroft C J, Beale M A, Breuer J. Clinical and biological insights from viral genome sequencing. Nature Reviews Microbiology, 2017, 15(3): 183-192.
- Li Y, Wang H, Nie K, et al. VIP: an integrated pipeline for metagenomics of virus identification and discovery. Scientific reports, 2016, 6.
Viral Metagenomic Seq Case Studies
Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study
Journal: The Lancet Infectious Diseases
Impact factor: 19.864
Published: February 17, 2016
Backgrounds
The microcephaly cases in Brazil in 2015 were 20 times more than in previous years. Epidemiological data suggest that the incidence of microcephaly in Brazil might be associated with Zika virus. The authors took amniotic fluid samples from two pregnant women in Brazil whose fetuses were diagnosed with microcephaly, in efforts to investigate the cause of microcephaly.
Methods
- Pregant women with symtoms of Zika virus infection
- Fetuses diagosed with microcephaly
- 5mL of amniotic fluid samples from two women at gestational week 28
- Serology tests
- ELISA
- Purification of viral particles
- Quantitative reverse transcription PCR
- Viral metagenomic sequencing
- PRINSEQ
- BLAST
- RefSeq
- De novo assembly
- Phylogenetic analysis
Results
1. Zika virus genome assembly
After removing cellular contamination, 288 904 sequences had similarity with virus genomes, and 683 sequences matched the Zika virus genome. Figure 1 shows the whole Zika virus genome isolated from patient 1 with gene annotation.
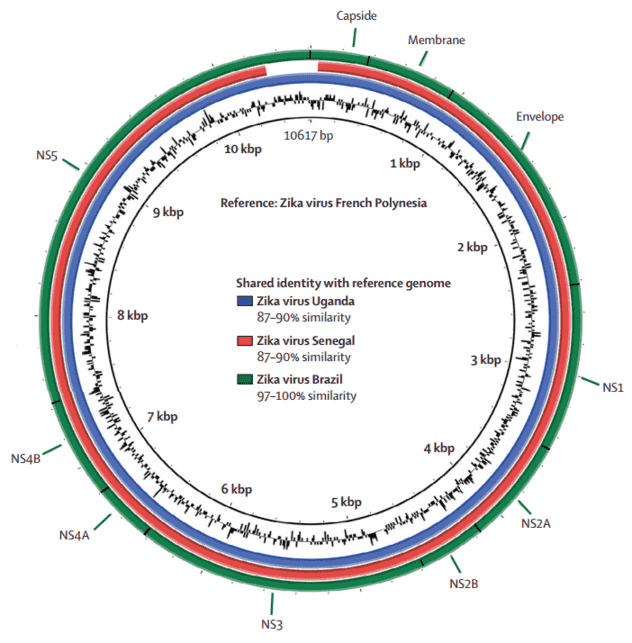 Figure 1. Comparative genome BLAST Atlas diagram of Zika virus. The green circle corresponds to the complete Brazilian Zika virus isolated from patient 1. The red circle corresponds to the Senegal strain of Zika virus. The blue circle corresponds to the Uganda strain.
Figure 1. Comparative genome BLAST Atlas diagram of Zika virus. The green circle corresponds to the complete Brazilian Zika virus isolated from patient 1. The red circle corresponds to the Senegal strain of Zika virus. The blue circle corresponds to the Uganda strain.
2. Phylogenetic analysis
The phylogenetic analyses were done with the coding region for envelope (Figure 2) and NS5 genes (Figure 3), respectively. The geographical region of the Brazilian Zika virus strain could not be inferred due to sampling limitations, but it seemed more closely related to French Polynesia than African strains.
The geographical and chronological distributions of Zika virus lineages indicated that southeast Asian strain could have genetically isolated from African strains for approximately 50 years. This pattern was further confirmed by the genetic distance between the new Brazilian Zika virus sequences and the Ugandan Zika virus genome (Figure 4).
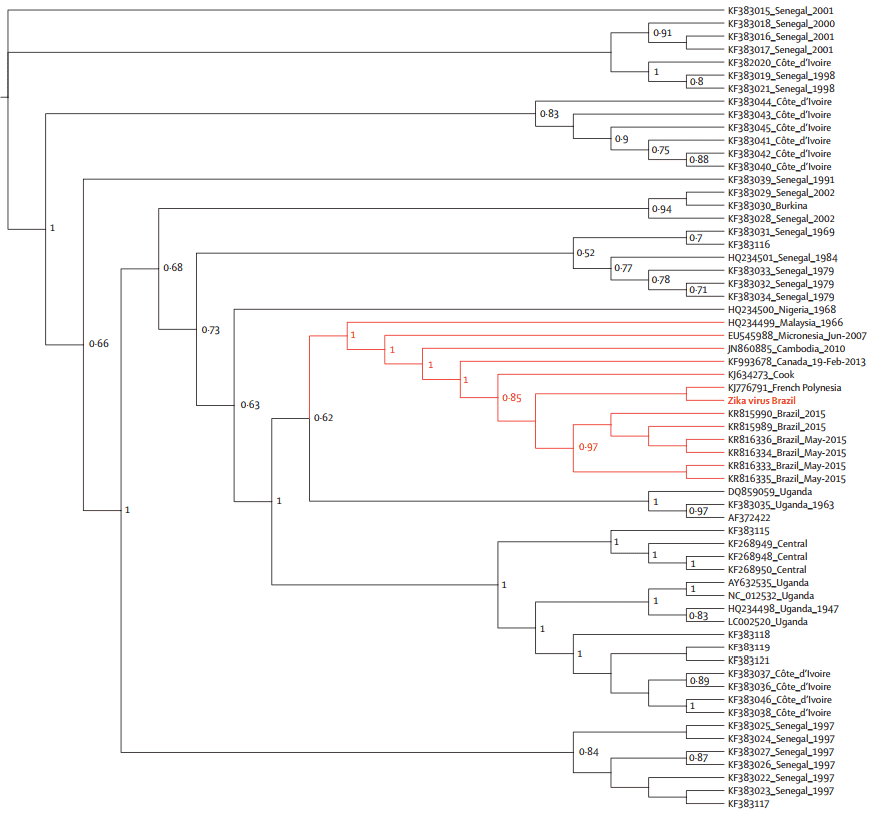 Figure 2. Maximum likelihood topologies of envelope genomic region from Brazilian Zika virus.
Figure 2. Maximum likelihood topologies of envelope genomic region from Brazilian Zika virus.
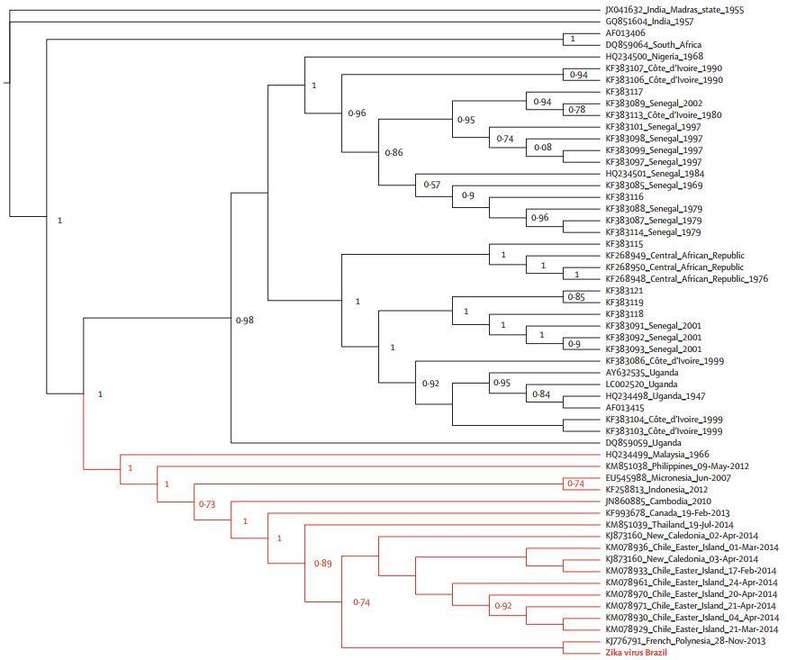 Figure 3. Maximum likelihood topologies of the NS5 genomic region from Brazilian Zika virus.
Figure 3. Maximum likelihood topologies of the NS5 genomic region from Brazilian Zika virus.
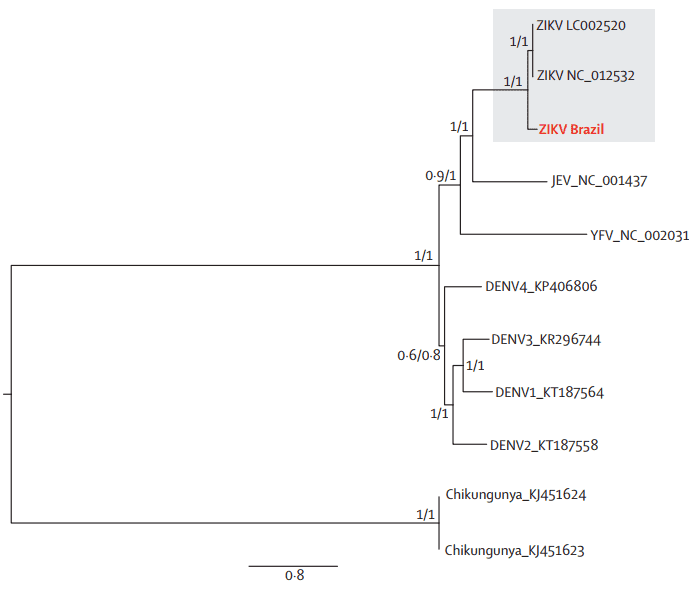 Figure 4. Maximum likelihood phylogeny of Brazilian Zika virus, other Flaviviridae genomes, and an alphavirus genome. DENV=dengue virus, JEV=Japanese encephalitis virus. YFV=yellow fever virus. ZIKA=Zika virus.
Figure 4. Maximum likelihood phylogeny of Brazilian Zika virus, other Flaviviridae genomes, and an alphavirus genome. DENV=dengue virus, JEV=Japanese encephalitis virus. YFV=yellow fever virus. ZIKA=Zika virus.
3. Zika virus infection could occur through the transplacental transmission
This article was the first to isolate the whole genome of Zika virus from amniotic fluid, which suggested that Zika virus infection could occur through the transplacental transmission.
Reference:
- Calvet G, Aguiar R S, Melo A S O, et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. The Lancet infectious diseases, 2016, 16(6): 653-660.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Transferrable protection by gut microbes against STING-associated lung disease
Journal: Cell Reports
Year: 2021
Microbial adaptation and response to high ammonia concentrations and precipitates during anaerobic digestion under psychrophilic and mesophilic conditions
Journal: Water Research
Year: 2021
Algal-bacterial synergy in treatment of winery wastewater
Journal: NPJ Clean Water
Year: 2018
Black soldier fly bioconversion to cultivated meat media components using blue catfish gut microbiome
Journal: Bioresource Technology Reports
Year: 2024
Indole-3-Propionic Acid, a Gut Microbiota Metabolite, Protects Against the Development of Postoperative Delirium
Journal: Annals of Surgery
Year: 2023
Elucidating the effects of organic vs. conventional cropping practice and rhizobia inoculation on rhizosphere microbial diversity and yield of peanut
Journal: Environmental Microbiome
Year: 2023
See more articles published by our clients.


