
 Sample Submission Guidelines
Sample Submission Guidelines
CD Genomics has been providing the flexible and affordable whole exome sequencing service for couple of years. We employ Illumina HiSeq sequencing platform to obtain the genetic variations information in a more efficient way.
WGS vs WES vs Targeted Panels: A Quick Method-Selection Guide
Not sure whether you need breadth, depth, or a focused gene list? The wrong assay can mean too much data, not enough depth, or missing key variant types. Use the flow below to match scope to your research question.
Quick Decision Flow
Q1. Do you need genome-wide discovery beyond coding regions, or is structural variation a primary focus?
- Yes → WGS
- No → Go to Q2
Q2. Is your goal broad discovery or screening of coding variants across many genes?
- Yes → WES
- No → Go to Q3
Q3. Do you already have a defined gene list / tight hypothesis?
- Yes → Targeted Panel
- No → WES
Q4. Do you need very high depth on a limited set of loci?
- Yes → Targeted Panel (or custom targeting)
- No → WES
Recommended Strategy
- Best balance for coding-variant discovery with manageable data volume.
- Ideal when you need discovery beyond a fixed gene list, without genome-wide complexity.
- Best for maximum breadth and projects needing genome-wide signals.
- Expect higher data volume and downstream analysis complexity.
Targeted Gene Panel Sequencing
- Best for a fixed gene list and high-depth, target-focused reads.
- Not intended for discovery outside the panel design.
1-Minute Comparison Table
| Decision factor | WGS | WES | Targeted Panel |
|---|---|---|---|
| Scope | Genome-wide | Protein-coding exome | Selected genes/regions |
| Best for | Maximum breadth | Coding discovery | Hypothesis-driven targets |
| Data scale | High | Medium | Low |
| Depth strategy | Broad | Balanced | Very high on targets |
| Interpretation effort | Higher | Moderate | Lower |
| Limits | More data handling | Limited outside exome | Limited outside panel |
Learn More
The Introduction of Whole Exome Sequencing
Human genome comprises approximately 3×109 bases, and contains approximately 180,000 coding regions (exome), constituting about 1.7% of a human genome. It is estimated that 85% of the disease-causing mutations occur in the exome. For this reason, sequencing of the whole exome has the potential to uncover higher yield of relevant variants at a far lower cost than whole genome sequencing. Whole exome sequencing is thought to be an efficient and powerful way to identify the genetic variants that affect heritable phenotypes, including important disease-causing mutations and natural variations that can be used to improve crops and livestock.
Whole Exome Sequencing utilizes exome capture technology to enrich exons, and then sequences these regions in a high-throughput manner. To be specific, DNA samples are first fragmented and biotinylated oligonucleotide probes (baits) are used to selectively hybridize to exome in the genome. Magnetic streptavidin beads are then used to bind to the biotinylated probes. The non-targeted portion of the genome is washed away, and the PCR is used to enrich the sample for DNA from the target region. Subsequently, the sample is sequenced by the Illumina HiSeq platform. This strategy can result in up to a 100-fold improvement in gene coverage for the human genome. The validated sequencing data are then used for variant analysis and research interpretation.
Our Exome Sequencing Solutions
At CD Genomics, we offer tailored Exome Sequencing services for Human/Mouse and Animal/Plant genomes, providing precise variant detection and cost-effective solutions.
| Service Type | Recommended Data Size | Notes |
|---|---|---|
| Human/Mouse Exome Sequencing | ||
| - Core Panel | ≥8 Gb @ 100X | Optimized for coverage and efficiency |
| - Inherited Panel | ≥11 Gb @ 100X | Enhanced SNV/InDel/CNV detection |
| - Tumor Panel | ≥20 Gb @ 200X | Supports TMB, MSI, and fusion detection |
| Animal/Plant Exome Sequencing | Varies by species | Targeted sequencing for various species (e.g., wheat, maize, cattle) |
Explore our Human/Mouse Exome Sequencing services or Animal/Plant Exome Sequencing options to find the perfect solution for your research.This concise solution helps you select the appropriate exome sequencing service for your research needs.
Advantages of Whole Exome Sequencing
- Lower cost and wide availability
- Increased sequence coverage (above 120X)
- Detection of coding single-nucleotide polymorphism (SNP) variants as sensitive as whole genome sequencing
- A smaller data set for faster and easier analysis compared to whole genome sequencing
- Medical and agricultural applications
Whole Exome Sequencing Workflow
CD Genomics employs the Illumina HiSeq system to provide the fast and accurate whole exome sequencing and bioinformatics analysis. Our highly experienced expert team executes quality management, following every procedure to ensure confident and unbiased results. The general workflow for whole exome sequencing is outlined below.

Sample Requirements
Project requirements may vary by species, capture design, and study goals. We'll confirm final input/QC thresholds during design review.
| Sample type | Recommended input (guideline) | QC suggestions | Notes |
|---|---|---|---|
| Genomic DNA (gDNA) | ≥ 500 ng | OD260/280 ~1.8–2.0; Qubit quantification; minimal degradation | Preferred for standard WES workflows |
| Blood / Cells / Fresh tissue (as DNA source) | As required to extract ≥ 500 ng gDNA | Provide extraction method info if available | We can advise on extraction if needed |
| FFPE tissue (as DNA source) | Project-dependent (often higher input) | DV200/fragmentation assessment recommended | Capture uniformity can be affected; plan QC conservatively |
| Low-input samples | Project-dependent | Pre-assessment recommended | Ask us for feasibility and targeting strategy |
What we check (typical): DNA quantity, purity, integrity/fragmentation (as applicable), library QC checkpoints (e.g., size distribution, complexity indicators).
Sequencing Strategy
1. Library & capture: Hybridization-based exome capture (typical target size 35–65 Mb).
2. Sequencing mode: Paired-end PE150 (150 bp × 2), dual-indexed libraries.
3. Coverage options (mean on-target depth):
- Standard WES: ~100×
- Deep WES: ~200×
- High-depth option: ~300× (project-dependent)
4. Approx. data per sample (raw):
- 100×: ~8–12 Gb
- 200×: ~15–22 Gb
- 300×: ~22–30 Gb
5. QC reporting: on-target performance, duplication, and coverage distribution (including ≥20× coverage summary across targets).
Bioinformatics Analysis
A standardized, reproducible bioinformatics pipeline to turn raw reads into interpretable variant and QC outputs.
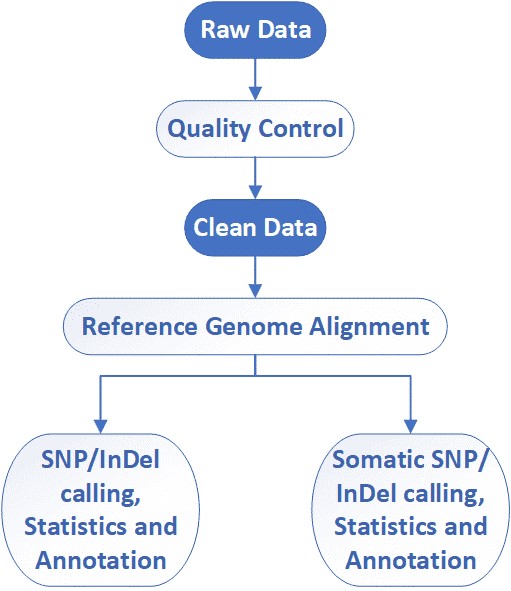
Core pipeline (included):
- Raw data QC:
- Alignment
- Coverage & capture QC
- Variant calling
- Variant annotation
- Summary reporting
Optional add-ons (choose as needed):
- Cohort-level joint analysis
- CNV inference from exome data
- Family-based analysis
- Custom filtering rules & prioritization aligned to your hypothesis/gene sets
- Downstream interpretation support
What You'll Receive (Deliverables)
Data files (analysis-ready):
- Raw reads: FASTQ (+ QC summaries)
- Aligned reads: BAM/CRAM (+ alignment metrics)
- Variant calls: VCF for SNVs/indels (and optional CNV outputs if selected)
- Annotated variant tables: TSV/Excel-style tables with configurable columns for filtering/prioritization
- QC & coverage package: key capture/coverage metrics, on-target and uniformity summaries, duplication and library indicators
Reports & summaries:
- Project report: concise overview of sample QC, sequencing/capture performance, and overall outcomes
- Variant summary: counts by type, distribution summaries, and cohort-level comparison snapshots (if cohort workflow is selected)
Reusable assets (for your team):
- Clear file naming + folder structure for pipeline integration
- Methods-ready description of the analysis steps for internal documentation and manuscript preparation (RUO)
CD Genomics provides full whole exome sequencing service package including sample standardization, exome capture, library construction, deep sequencing, raw data quality control, and bioinformatics analysis. We can tailor this pipeline to your research interest. If you have additional requirements or questions, please feel free to contact us.
Reference:
- Warr A, Robert C, Hume D, et al. Exome sequencing: current and future perspectives. G3: Genes, Genomes, Genetics, 2015, 5(8): 1543-1550.
Demo Results
These example plots highlight typical WES outputs—coverage/QC performance, variant summary, enrichment overview, and genome-wide signal visualization—for quick project review.
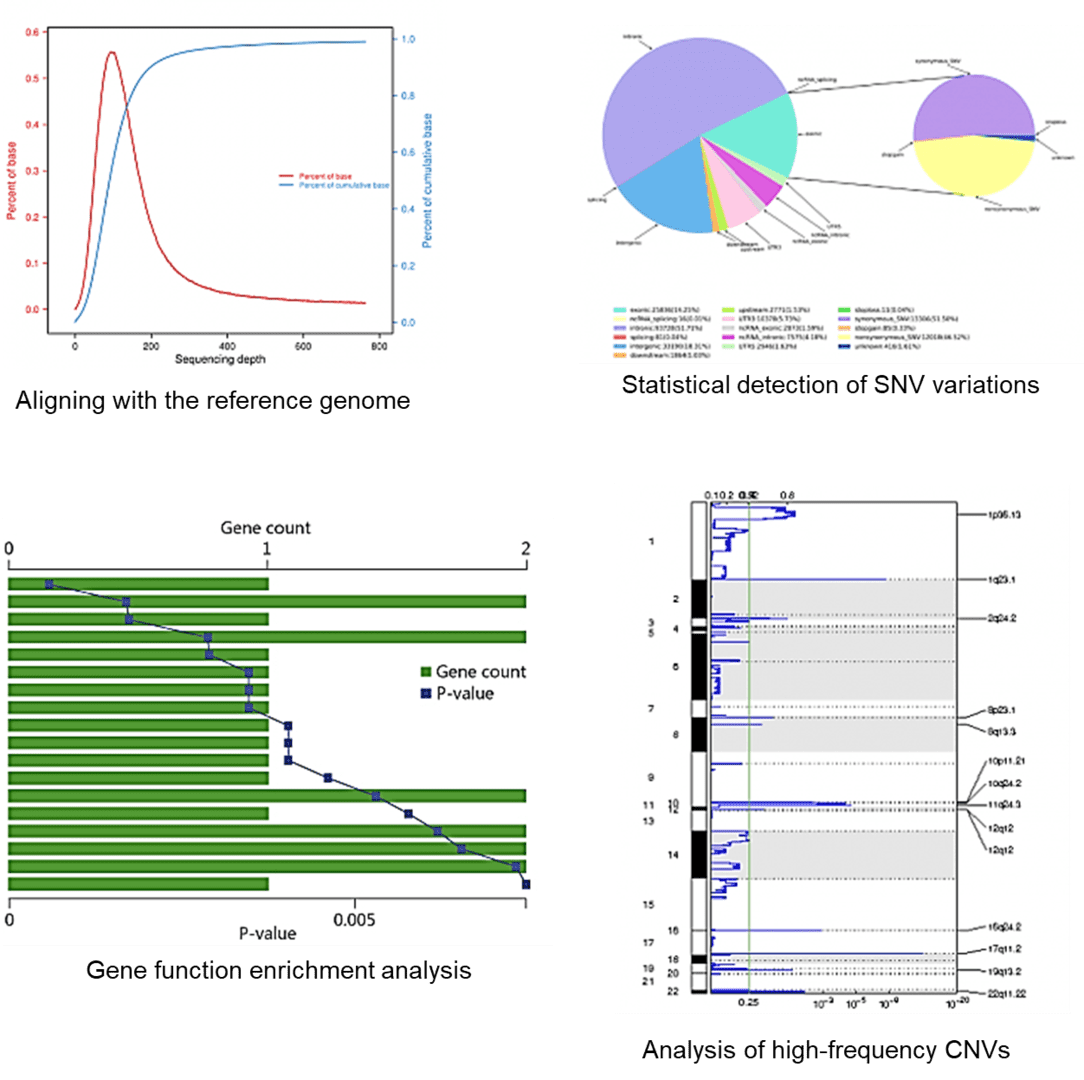
Whole Exome Sequencing FAQs
1. What are the applications of whole exome sequencing?
Human genome contains approximately 180,000 coding regions (exome), constituting about 1.7% of a human genome. It is estimated that 85% of the disease-causing mutations occur in the exome. Therefore, whole exome sequencing is a potential contributor to the understanding of human diseases. Whole exome sequencing is a cost-effective and powerful tool, especially suitable for bigger sample size and high coverage. Whole exome sequencing is mainly used to investigate the genetic cause of both Mendelian and common diseases such as cancer and diabetes.
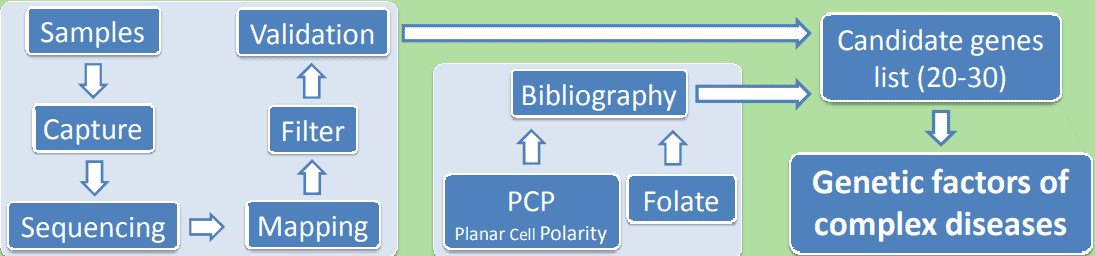 Figure 1. Application of whole exome sequencing in a complex disease.
Figure 1. Application of whole exome sequencing in a complex disease.
2. What variations can whole exome sequencing detect?
Whole exome sequencing can detect SNPs, InDels, and maybe copy number variations (CNVs).
3. How do I determine the sequencing depth?
Sequencing depth is an important factor for high-throughput sequencing. One paper published in the journal Genomics & Informatics revealed that the sequencing depth of whole exome sequencing can affect the discovery rates of variations. To summarize, the number of deleterious SNPs and InDels detected in the coding regions was only weakly increased a depths more than 120×. In other words, a sequencing depth of 120× can be considered reasonable when using the exome capture sequencing technique to identify significant variations in diagnostic studies.
4. What are the disadvantages of whole exome sequencing?
Whole exome sequencing is characterized by lower cost, increased sequence coverage, as well as sensitive and specific identification. Nevertheless, Whole exome sequencing cannot detect structural variants, and has a limited view, i.e., only coding regions. Not all targets are captured (approximately 80%), and it is difficult to capture GC-rich regions.
5. What's the difference between WES and WGS?
Whole exome sequencing (WES) targets protein-coding regions for efficient variant discovery with manageable data volume, while WGS profiles the entire genome for maximum breadth and re-mining potential.
6. What regions does whole exome sequencing cover?
WES focuses on exome capture of protein-coding regions (target size typically in the tens of megabases, depending on the capture design), with coverage varying across GC-rich or hard-to-capture targets.
7. What QC metrics are reported for WES?
We report key coverage and capture QC such as on-target performance, duplication, and coverage distribution (including common coverage threshold summaries across targets) to help you assess data usability quickly.
8. What files will I receive from a WES project?
Typical deliverables include FASTQ (raw reads), BAM/CRAM (aligned reads), VCF (variant calls), plus an annotated variant table and a concise QC/report package (RUO).
9. Can WES results be used for cohort-level comparison?
Yes. For multi-sample studies, outputs can be generated in a consistent format and (optionally) with cohort-aware analysis to improve comparability across samples and reduce batch-related interpretation issues.
Reference:
- Kyung Kim, et al. Effect of Next-Generation Exome Sequencing Depth for Discovery of Diagnostic Variants. Genomics & Informatics. 2015, Jun; 13(2): 31–39.
Whole Exome Sequencing Case Studies
First missense mutation outside of SERAC1 lipase domain affecting intracellular cholesterol trafficking
Journal: Nuerogenetics
Impact factor: 3.269
Published online: 7 October 2015
Abstract
MEGDEL syndrome is a rare inborn error of metabolism. This syndrome has been associated to mutations in the serine active site containing 1 (SERAC1) gene. The authors reported a new homozygous mutation in the SERAC1 gene via whole exome sequencing at CD Genomics. This is the first missense mutation outside of serine-lipase domain of the protein that affects the intracellular cholesterol trafficking.
Results
1. Mutations in the SERAC1 gene
To date, 19 mutations in the SERAC1 gene have been identified in patients with MEGDEL syndrome (Table 1). Only three are missense mutations which are localized within the lipase domain. The p.D224G is the first missense mutation outside of lipase domain.
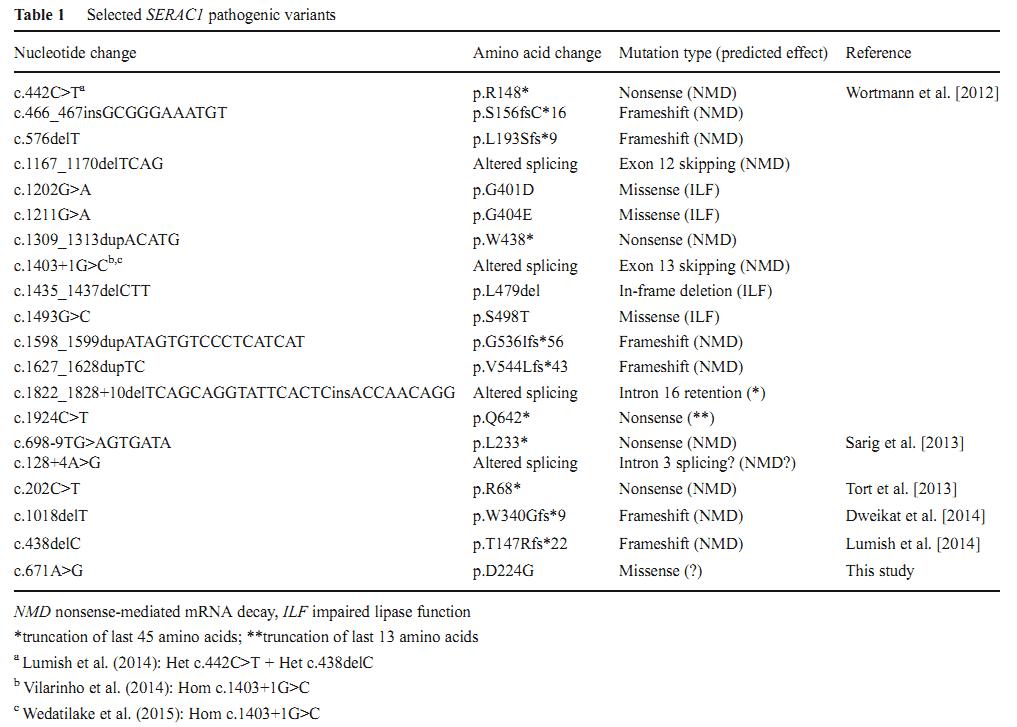
2. Missense mutation (p. D224G)
Using whole exome sequencing, the authors identified a novel pathogenic homozygous mutation in the SERAC1 gene. This missense mutation changed a aspartic acid to glycine (Figure 1d). The pathogenic role of p.D224G is supported by in silico analysis, the conservation of the mutant amino acid residue (Figure 1d), and the accumulation of cholesterol (Figure 1e).
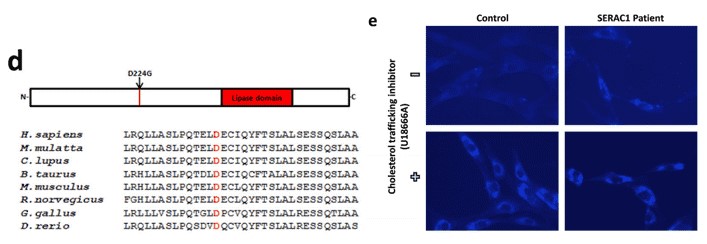 Figure 1. The position of D224 mutation in various species (d). Intracellular cholesterol trafficking in fibroblasts derived from healthy individual and the SERAC1 patients. U1866A is an inhibitor of cholesterol trafficking.
Figure 1. The position of D224 mutation in various species (d). Intracellular cholesterol trafficking in fibroblasts derived from healthy individual and the SERAC1 patients. U1866A is an inhibitor of cholesterol trafficking.
Reference:
- Rodríguez-García M E, et al. First missense mutation outside of SERAC1 lipase domain affecting intracellular cholesterol trafficking. Neurogenetics, 2016, 17(1): 51-56.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Optical Genome and Epigenome Mapping of Clear Cell Renal Cell Carcinoma
Journal: bioRxiv
Year: 2022
An independent origin of an annual life cycle in a North American killifish species
Journal: Biological Journal of the Linnean Society
Year: 2024
Combinations of Bacteriophage Are Efficacious against Multidrug-Resistant Pseudomonas aeruginosa and Enhance Sensitivity to Carbapenem Antibiotics
Journal: Viruses
Year: 2024
Genome sequence, antibiotic resistance genes, and plasmids in a monophasic variant of Salmonella typhimurium isolated from retail pork
Journal: Microbiology Resource Announcements
Year: 2024
High-Density Mapping and Candidate Gene Analysis of Pl18 and Pl20 in Sunflower by Whole-Genome Resequencing
Journal: International Journal of Molecular Sciences
Year: 2020
Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI
Journal: New phytologist
Year: 2017
See more articles published by our clients.


