
 Sample Submission Guidelines
Sample Submission Guidelines
Full-Length 16S/18S/ITS Amplicon Sequencing
In terms of the extensive experience in sequencing, CD Genomics is proud to offer our clients full-length 16S/18S/ITS rRNA services with the best quality and most competitive price.
What Is Full-length 16S/18S/ITS Amplicon Sequencing
The 16S rRNA gene is highly conserved between different species of bacteria and archaea, which contains nine hypervariable regions (V1-V9) ranging from about 30~100 base pairs long, they vary dramatically between bacteria. Highly conserved regions allow researchers to design primer pairs that will accurately and reliably amplify the 16s hypervariable region of their choice to achieve identification or characterization of diverse bacterial communities.
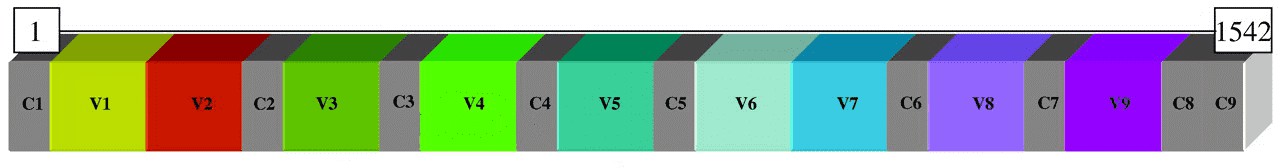
Similar to the bacterial 16S rRNA genes, the eukaryotic 18S rRNA gene has conserved and variable regions. 18S rRNA gene sequences and their associated transcribed spacers (internal transcribed spacer; ITS) are used to classify fungi and eukaryotes.
Based on the development of sequencing technology, 16S/18S/ITS rRNA gene sequencing is the best tool to study bacterial and fungal taxonomy and molecular phylogeny. By taking advantage of PacBio SMRT long reads sequencing technology, CD Genomics can provide full-length 16S/18S/ITS rRNA sequencing service to better help your research.
Full-length sequencing of 16S/18S/ITS entails the extraction of microbial DNA from a given sample, followed by the amplification of the microbial 16S rDNA, 18S rDNA, or ITS region in its entirety using universal primers, and subsequent sequencing. This methodology not only boosts the precision of species identification, thanks to improved resolution, but also enhances the accuracy in deciphering the microbial constitution within samples. Consequently, this offers a more integrative insight into the structure of the microbial community.
With such heightened resolution, the focus naturally shifts to species-level studies. This represents a significant progression from previous second-generation studies centered on the genus and species level using 16S. Third-generation full-length 16S can provide more comprehensive and intricate strain-level analysis, rendering the overall research outcomes more relevant to ecological functionality. This approach greatly impacts multi-omics correlations and subsequent experimental guidelines and validations, giving it significant meaning. From the perspective of multi-omics correlation, finer-level data often reveals clearer local patterns, encompassing many details that were previously either overlooked or inaccessible.
Advantages of Full-length 16S/18S/ITS Amplicon Sequencing
- Longer reads for increased accuracy.
- Higher species identification resolution and precision.
- More accurate reconstruction of microbial communities.
- Mature sequencing platform: PacBio SMRT system.
- High-quality data assurance.
- Accurate identification with multiple options for CCS reads.
- Rich project experience.
- Diverse analysis content.
- Short cycles with competitive pricing advantages.
Applications of Full-length 16S/18S/ITS Amplicon Sequencing
- Medical field: Research on the relationship between common diseases and human microbiota.
- Animal field: Studies on the relationship between the gut, rumen (such as methane-producing microbial communities), and animal health/nutrient digestion.
- Agricultural field: Research on rhizosphere microbiota and plant interactions, as well as agricultural cultivation/fertilization practices and soil microbial communities.
- Environmental field: Investigations into haze treatment, wastewater management, petroleum degradation, acid mine drainage treatment, and marine environment research.
- Special extreme environments: Research on microbial communities under extreme environmental conditions.
Workflow of Full-length 16S/18S/ITS Amplicon Sequencing
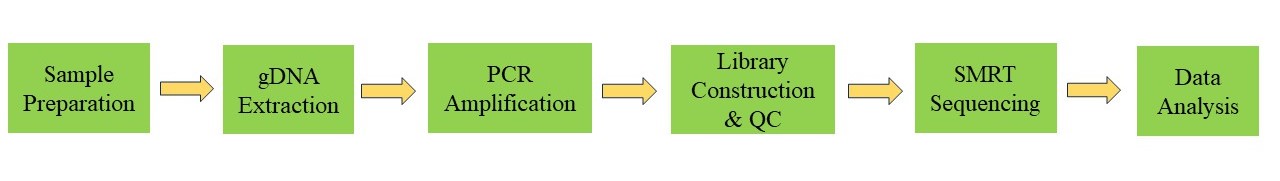
Service Specification
Sample Requirements:
|
|
 |
Sequencing:
|
 |
Data Analysis
|
Bioinformatics Pipeline

Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Full-Length 16S/18S/ITS Amplicon Sequencing for your writing (customization)
References:
- Callahan B J, Wong J, Heiner C, et al. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic acids research, 2019, 47(18): e103-e103.
- Lam T Y C, Mei R, Wu Z, et al. Superior resolution characterisation of microbial diversity in anaerobic digesters using full-length 16S rRNA gene amplicon sequencing. Water Research, 2020, 178: 115815.
- Hui M, Wang A, Cheng J, et al. Full-length 16S rRNA amplicon sequencing reveals the variation of epibiotic microbiota associated with two shrimp species of Alvinocarididae: possibly co-determined by environmental heterogeneity and specific recognition of hosts. PeerJ, 2022, 10: e13758.
Demo Results
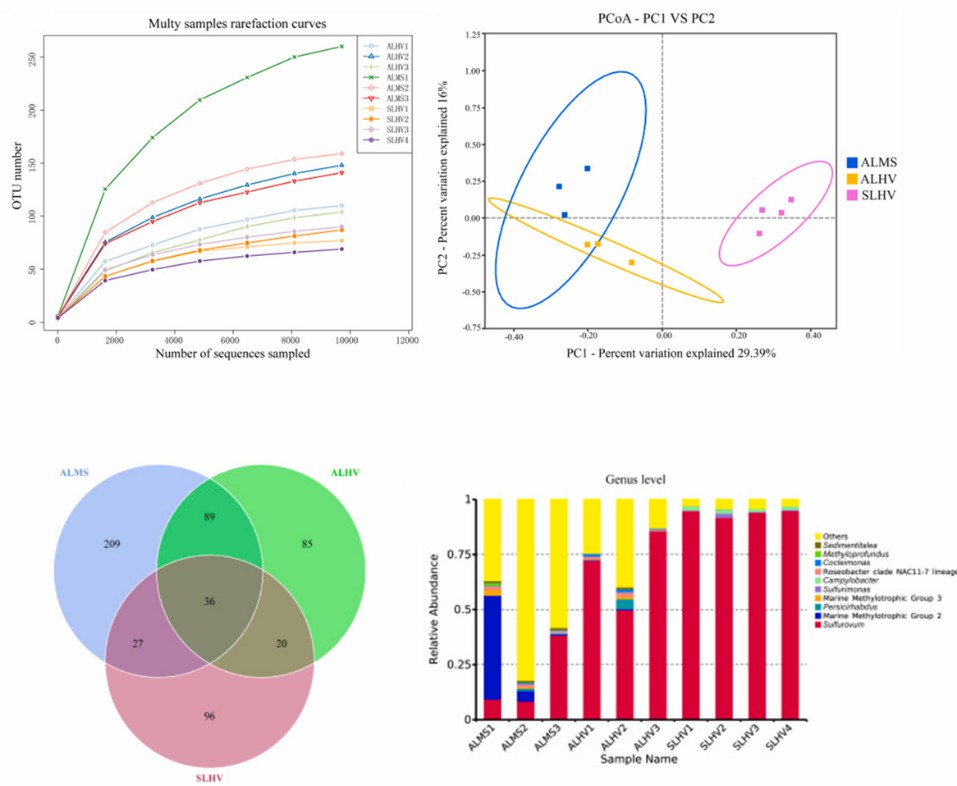 Using the results of 16S rRNA amplicon sequencing for display. (Hui, et al., 2022)
Using the results of 16S rRNA amplicon sequencing for display. (Hui, et al., 2022)
Full-Length 16S/18S/ITS Amplicon Seq FAQs
1. What are the advantages of third-generation amplicon sequencing?
Third-generation amplicon sequencing offers several advantages. Leveraging the PacBio SMRT single-molecule sequencing technology, it provides ultra-long reads, enabling the coverage of full-length ribosomal small subunit 16S/18S/ITS regions. This addresses the technical challenge of short-read products, which are limited to analyzing only local variable regions. Moreover, it achieves truly precise microbial classification and identification at reasonable costs.
2. Why choose full-length microbial diversity?
The second-generation sequencing technique primarily analyses partial sequences of the 16S rRNA gene. However, different researchers analyze different regions of 16S rRNA genes in diverse bacteria, and the choice of PCR primers for short-read amplicons of different hypervariable regions can influence the inferred accuracy of the community and the sensitivity to certain bacterial groups. This situation renders comparisons on a global level in microbiome studies challenging. Additionally, due to the limited mutation information carried by some hypervariable regions, species-level annotation cannot be achieved. Therefore, the diversity of second-generation microbiomes is typically studied at the genus level and above. In contrast, third-generation sequencing can measure all the hypervariable regions in one go. This capability eliminates the preferences brought about by varying primers and offers a novel method for improving the taxonomic resolution of microbial communities.
3. How to identify bacterial species?
Compare obtained 16S rDNA sequences to reference 16S rDNA sequences on GenBank or Eztaxon. With more than 97% of sequence similarity, they can be considered as the same bacterial species. For more accurate identification, DNA hybridization, genomic GC content, and physiological and biochemical indexes should be used.
4. The requirements for samples.
If you submit cultured microbes, please make sure they have been purified for at least three times. And please give a clear indication of the cultural conditions. If any special medium is needed, please provide it.
If you submit genomic DNA samples, the amount with more than 500 ng are required.
For Gram-positive microbes, please submit genomic DNA samples.
5. What is the full length of the 16S/18S/ITS sequence?
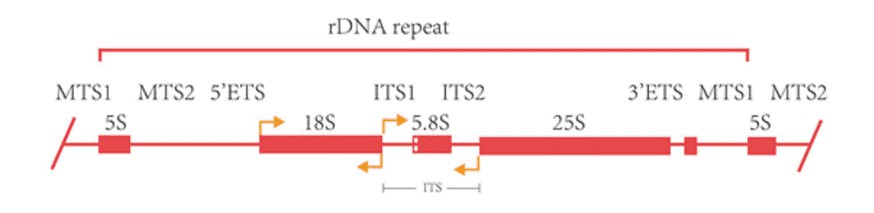
The 16S rRNA gene of prokaryotic microbes approximates 1500 bp in length, encompassing ten conserved regions and nine highly variable regions (V1-V9). It exhibits both a high level of conservativeness and specificity, with the conserved regions reflecting phylogenetic relationships among genera, and the variable regions depicting inter-genus variation. The 18S rRNA gene of eukaryotic microbes, with a sequence length ranging between 1500-2000 bp, also features both conserved and variable regions. Due to its conservative evolution rate, it is suitable for taxonomic identification at the species level and above. The internal transcribed spacer (ITS) of eukaryotic microbes, approximately 500 bp in length, retains high conservativeness among different strains within a species but undergoes considerable variation among species. This renders it extremely polychromatic, proving its wide application in inter-species phylogenetic studies and research.
Full-Length 16S/18S/ITS Amplicon Seq Case Studies
Analysis of the mouse gut microbiome using full-length 16S rRNA amplicon sequencing
Journal: Scientific reports
Impact factor: 4.6
Published: 14 July 2016
Abstract
The advent of next-generation sequencing technology has bolstered detailed clarification of microbial community composition, boasting enhanced accuracy and throughput compared to previous methods. The authors harnessed nanopore sequencing platforms to sequence a library of full-length 16S rRNA amplicons prepared from the gut microbiota of mice. At the species level, nanopore sequencing outperforms short read sequencing in the identification of more species, thereby contributing to an accurate characterization of bacterial community composition.
Methods
- Microbial metagenomic DNA
- Full-length 16S rDNA amplicon sequencing
- Illumina sequencing
- OTU taxonomy
- Sequencing accuracy calculated
Results
1. Statistical comparison between short-read and nanopore sequencing data
The short-read sequencing dataset was analyzed using the Operational Taxonomic Unit (OTU) approach. On the other hand, the composition of the microbial community was determined based on Nanopore sequencing data obtained through a systematization method, known as a taxonomically supervised analysis, which directly allocates sequences into taxonomic bins based on sequence similarity. Despite the negligible level of sample-specific phylogeny observed and the relatively high error rate (20.4%) identified from the Nanopore sequencing reads, high reproducibility and correlation were achieved between biological replicates. Consequently, the application of Nanopore amplicon sequencing allows for the repeated detection of 34 main system types.
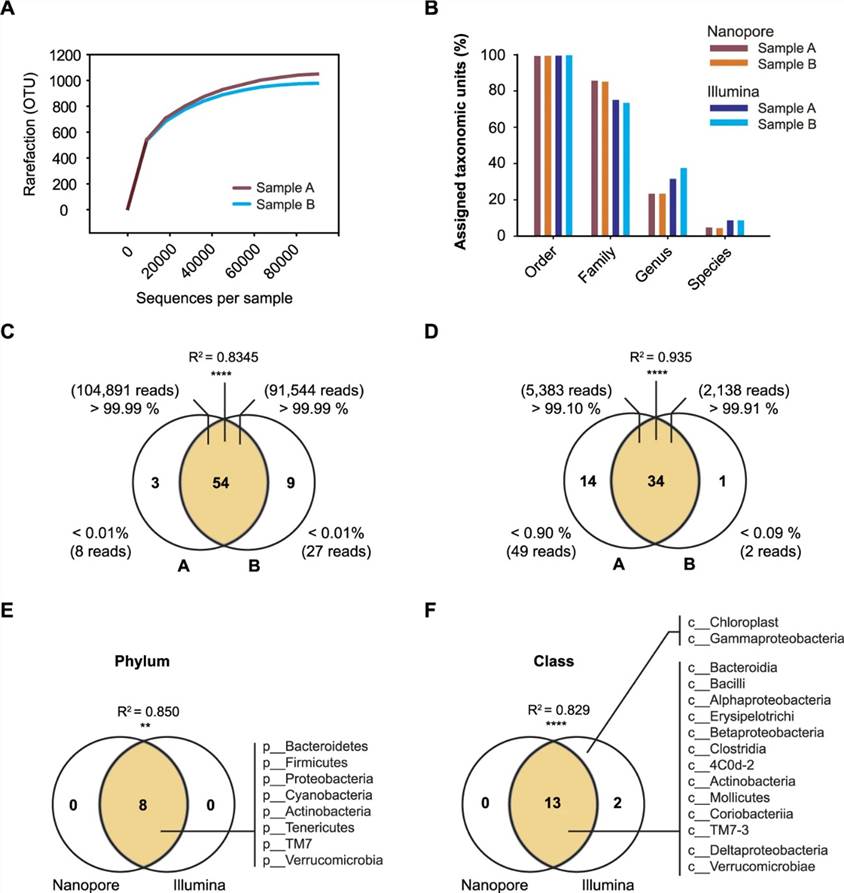 Figure 1. Statistical comparison between short-read and nanopore sequencing data.
Figure 1. Statistical comparison between short-read and nanopore sequencing data.
2. Comparison of microbial composition determined by two sequencing platforms
The authors compared the microbial composition determined using two sequencing platforms. Both platforms identified 8 bacterial phyla and 13 bacterial classes (as illustrated). There was notable statistical similarity between short-read and nanopore sequencing regarding the relative proportions and classes of principal phyla. In general, significant similarities in bacterial composition were observed between the two platforms at the order, family, and species level. From comparative analysis of microbial composition independently analyzed via nanopore and short-read sequencing platforms, it is evident that nanopore sequencing can accurately identify microbial composition at a species level.
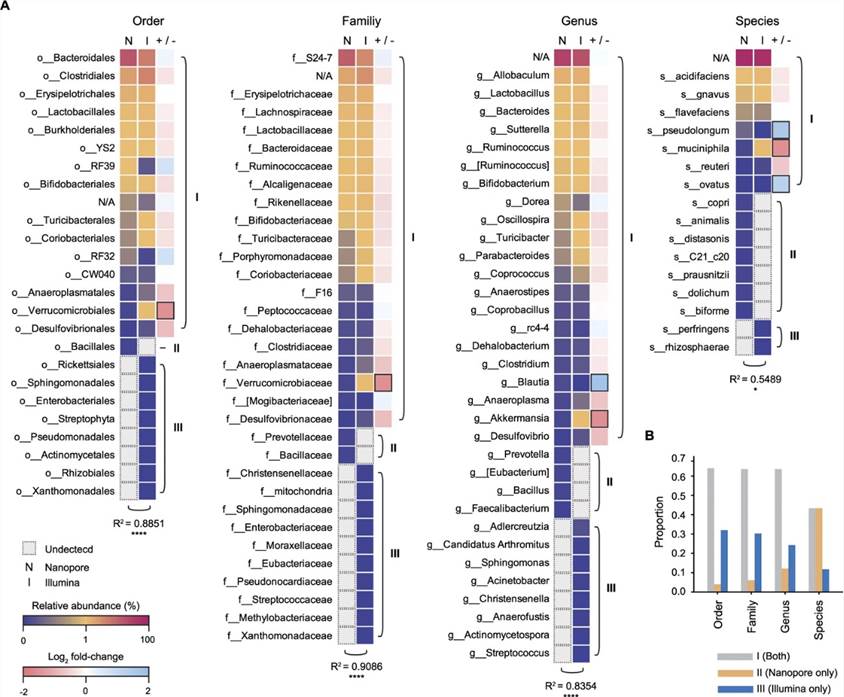 Figure 2. Comparison of mouse gut microbiota compositions between two sequencing platforms at deeper classifications (order to species).
Figure 2. Comparison of mouse gut microbiota compositions between two sequencing platforms at deeper classifications (order to species).
3. Species detection using full-length 16S rDNA amplicon sequencing
In relation to the taxonomic resolution from phylum to species level, the relative proportions of Group I (both sequencing platforms) and Group III (only short-read sequencing) have declined, whereas Group II (nanopore sequencing only) has seen an increase. In this regard, we postulate that the long reads generated by nanopore sequencing cross multiple hypervariable regions, and due to its near full-length 16S rDNA reads, may outperform Illumina sequencing in discerning the microbial community structure. To determine the impact of longer reads on species-level microbial community analysis, we performed a phylogenetic analysis on the composite dataset based on priority and identified 16 phylogenetically distinct species distributed across 13 genera(as illustrated).
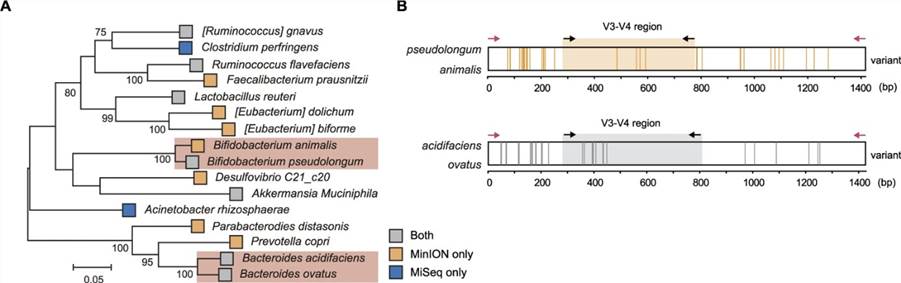 Figure 3. Phylogenetical analysis of mouse gut microbiota.
Figure 3. Phylogenetical analysis of mouse gut microbiota.
Conclusion
The authors explored the potential of the third-generation sequencer, MinION, in identifying microbial community composition in mouse fecal samples. Employing nanopore sequencing for full-length 16S rRNA amplicon sequencing, they were able to swiftly, accurately and efficiently determine microbial diversity at the species level. The success of this study in clarifying microbial community composition is indicative of the future potential of the method. The authors anticipate that this approach will expand the applicability of microbial community analyses across biological, clinical, and environmental origins.
Reference:
- Shin J, Lee S, Go M J, et al. Analysis of the mouse gut microbiome using full-length 16S rRNA amplicon sequencing. Scientific reports, 2016, 6(1): 29681.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Bacterial communities of Cassiopea in the Florida Keys share major bacterial taxa with coral microbiomes
Journal: bioRxiv
Year: 2024
Production of a Bacteriocin Like Protein PEG 446 from Clostridium tyrobutyricum NRRL B-67062
Journal: Probiotics and Antimicrobial Proteins
Year: 2024
Untangling the Role of Pathobionts from Bacteroides Species in Inflammatory Bowel Diseases
Journal: bioRxiv
Year: 2023
A chromosome-level genome resource for studying virulence mechanisms and evolution of the coffee rust pathogen Hemileia vastatrix
Journal: bioRxiv
Year: 2022
Streptomyces buecherae sp. nov., an actinomycete isolated from multiple bat species
Journal: Antonie Van Leeuwenhoek
Year: 2020
See more articles published by our clients.


