
 Sample Submission Guidelines
Sample Submission Guidelines
Small RNA Sequencing
To support the increasing research interest in small RNA, CD Genomics is offering the qualified small RNA sequencing service that covers novel small RNA discovery, mutation characterization, and expression profiling of small RNAs by leveraging of advanced NGS technologies and data analysis pipeline.
The Introduction of Small RNA Sequencing
Small RNA species generally include the most common and well-studied microRNA (miRNA), small interfering RNA (siRNA), and piwi-interacting RNA (piRNA), as well as other types of small RNA, such as small nucleolar RNA (snoRNA) and small nuclear RNA (snRNA). Small RNA is a type of lowly abundant, short in length (<200 nt), non-protein-coding RNAs that lack polyadenylation. Small RNA populations can vary significantly among different tissue types and species. Generally, small RNAs are formed by fragmentation of longer RNA sequences with the help of dedicated sets of enzymes and other proteins.
Small RNAs act in gene silencing and post-transcriptional regulation of gene expression. However, small RNA is not sufficient for the induction of RNA inference. It generally needs to form the core of the RNA-protein complex known as RNA-induced silencing complex (RISC). siRNAs can cleave the mRNA in the middle of the mRNA-siRNA duplex, and the resulting mRNA halves are degraded by other cellular enzymes. Unlike the siRNA pathway, miRNA-mediated degradation is initiated by enzymatic removal of the mRNA polyA tail. piRNAs are essential for the development of germ cells. Small RNAs have been demonstrated to be involved in a number of biological processes including development, cell proliferation and differentiation, and apoptosis.
By taking advantage of tremendous output with unprecedented sensitivity and dynamic range, NGS can identify weakly expressed small RNAs as well as quantitatively reveal heterogeneity in length and sequence. NGS is a powerful tool for investigating the function of small RNAs and prediction of potential mRNA target molecules without requiring available reference genomes. Obtaining a premium small RNA sequencing library begins with the isolation of small RNAs by size fractionation using gel electrophoresis selection or silica spin columns from total RNA. Following RNA adapter ligation using a 5' adenylated DNA adapter with a blocked 3'end, small RNAs are reverse transcribed, amplified by PCR and sequenced. To identify and annotate known miRNAs, the sequencing reads can be mapped to a species-specific database, such as mirWalk and miRBase.
Advantages of Our Small RNA Sequencing Service
- Small RNA and miRNA profiling
- Understanding how post-transcriptional regulation contributes to the phenotype
- Identifying more unmapped small RNAs and isoforms, as well as novel biomarkers
- High Resolution, High Accuracy: Detection of single-nucleotide differences, accurately counting from a few to tens of thousands of copies
- High Throughput, High Quality: Obtaining over 8 million sequences in one sequencing run, short experimental period, high efficiency, low cost, and reliable quality
- Standardized Analytical Workflow: Professional bioinformatics teams and analysis services to provide standardized, advanced, and customized analyses
- Extensive experience in small RNA sequencing and analysis: Professional sequencing technicians assisting in experimental design, problem-solving, and result analysis
- Customized small RNA sequencing protocols: Tailoring personalized small RNA sequencing plans according to specific needs.
Small RNA Sequencing Workflow
CD Genomics utilizes the Illumina HiSeq platforms to sequence small RNAs. We have flexible strategies for miRNA (15-30nt) and/or small RNA (30-200nt) discovery and profiling. Our highly experienced expert team executes quality management, following every procedure to ensure confident and unbiased results. The general workflow for small RNA sequencing is outlined below.

Service Specification
Sample Requirements
|
|
 Click |
Sequencing Strategies
|
 |
Data Analysis We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
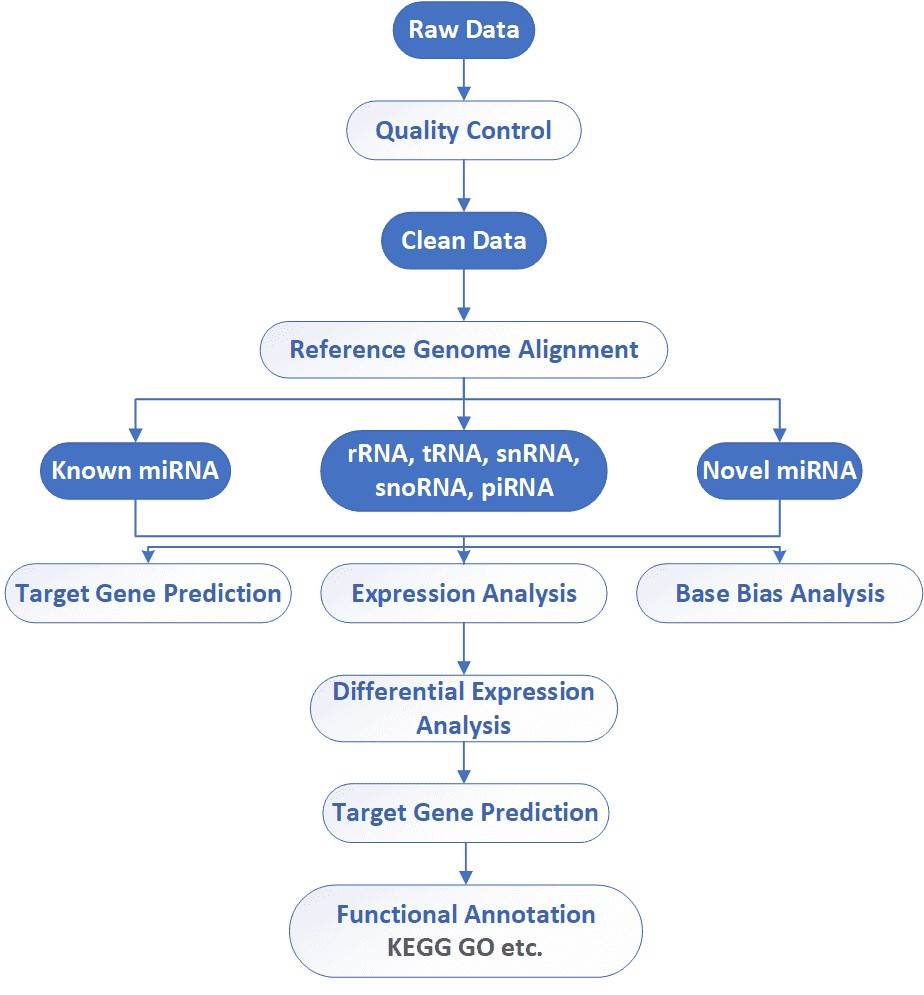
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Small RNA Sequencing for your writing (customization)
CD Genomics provides full small RNA sequencing service packages including sample standardization, library preparation, deep sequencing, raw data quality control, genome assembly, and customized bioinformatics analysis. We can tailor this pipeline to your research interest. If you have additional requirements or questions, please feel free to contact us.
Demo Results
Partial results are shown below:

Sequencing quality distribution

A/T/G/C Distribution

IGV Browser Interface

Correlation Analysis Between Samples

PCA Score Plot

Venn Diagram

Volcano Plot

Statistics Results of GO Annotation

KEGG Classification
Small RNA Seq FAQs
1. What is the workflow for data pre-processing?
The first steps are adapter trimming using FASTX and an optional k-mer correction using ECHO. Subsequently, the expression profile is formed by genome mapping and annotation. If the number of reads per sample varies, normalization is applied to offset under-sampling effects.
2. How many reads are required for small RNA sequencing?
It depends on your application. For expression profiling, an accepted range for mapped reads per sample is 100K-2M. For discovery applications, at least 5-10M should be considered.
3. How many biological replicates do I need for each condition?
Biological replicates can increase confidence and reduce experimental error, and we recommend you submit at least three replicates per sample. Note that the final number of replicates is determined by your final experimental conditions.
4. How should I ship samples?
You may ship with dry ice or RNAstable at room temperature. Empirically, shipping RNA samples in RNAstable yields good RNA quality after reconstitution.
5. Can I submit E. coli cells or tissue samples?
Yes, apart from RNAs, you can also submit frozen E. coli cell pellets or tissues with dry ice.
6. Is poly-A enrichment or RNA depletion required for microRNA sequencing?
No enrichment is required for microRNA sequencing because adapters are selectively ligated to small RNAs only.
7. I found that I could not open the downloaded fastq file.
Please make sure that your computer has enough RAM to open the large files. You can open them by text editor on windows computers with enough RAM, or on Linux computers, which is a preferred option. Alternatively, you may consider other fastq specific readers.
Small RNA Seq Case Studies
Novel microRNA discovery using small RNA sequencing in post-mortem human brain
Journal: BMC Genomics
Impact factor: 3.729
Published: 4 October 2016
Background
MicroRNAs (miRNAs) regulate gene expression primarily through translational repression of target mRNA molecules. Over 2700 human miRNAs have been identified and some were associated with disease phenotypes and showed tissue-specific patterns of expression. The authors conducted small RNA sequencing studies on 93 human post-mortem prefrontal cortex samples from patients with Huntington's disease or Parkinson's disease. And they successfully discovered 99 putative novel miRNAs.
Materials & Methods
- Frozen brain tissue
- Isolation and purification of total RNA
- Library construction
- Single-end sequencing
- Illumina's HiSeq 2000 system
- miRNA sequencing
- Small RNA Sequencing
- Identification of novel miRNAs
- Sequence similarity with known miRNAs
- Differential expression analysis
Results
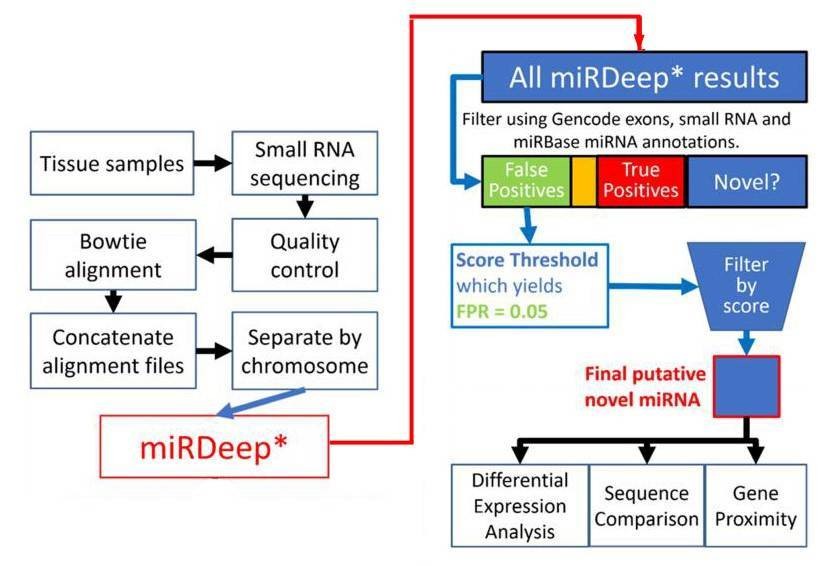 Figure 1. Flowchart depiction of the novel miRNA discovery pipeline.
Figure 1. Flowchart depiction of the novel miRNA discovery pipeline.
1. Total 8891 miRNAs were identified.
miRDeep* analysis of sequence data from 93 human prefrontal cortex samples identified 8891 miRNAs. 3641 miRNAs were remained after filtering for known miRNAs from miRBase v20, other small RNAs and exons. The rest were filtered by the miRDeep* score. Higher scores indicate higher confidence of a miRNA result. Total 99 putative novel miRNAs were finally identified.
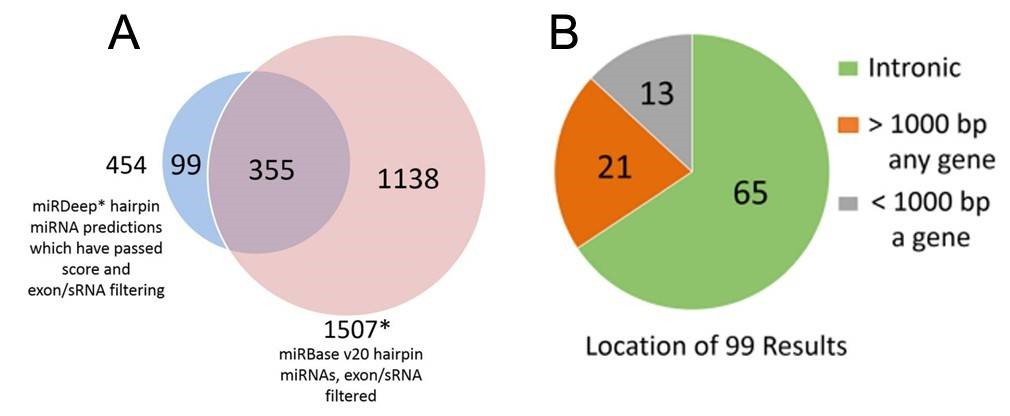 Figure 2. miRDeep* putative miRNAs and miRBase miRNAs (A), and genomic locations of the putative novel miRNAs (B).
Figure 2. miRDeep* putative miRNAs and miRBase miRNAs (A), and genomic locations of the putative novel miRNAs (B).
2. Total 99 putative novel miRNAs
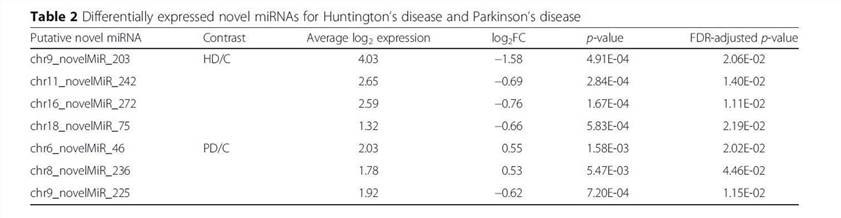
Using the SSEARCH aligner, 34 of the 99 putative novel mature miRNAs aligned well to at least one mature miRNA present in the miRBase v20. Differential expression analysis using linear models adjusting for age of death showed that 4 of the 99 putative miRNAs are differentially expressed between control and Huntington's disease samples, and that 3 were differentially expressed between control and Parkinson's disease samples (Table 2). The detailed data are presented in Table 2 that are generated by LIMMA v. 3.33.7.
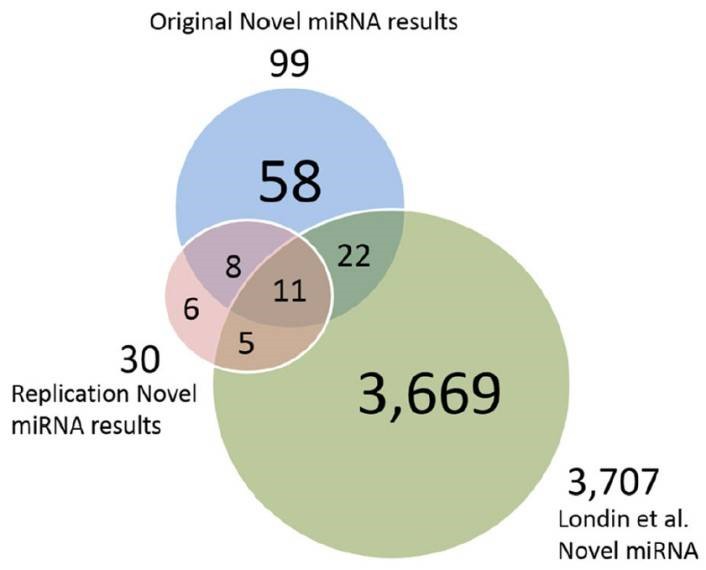 Figure 3. Putative novel miRNAs, replication data miRNAs and Londin et al. miRNAs.
Figure 3. Putative novel miRNAs, replication data miRNAs and Londin et al. miRNAs.
Conclusion
The authors developed a pipeline using miRDeep*, pooled RNA sequencing, and score filtering to discover 99 putative novel miRNAs from 93 post-mortem human prefrontal cortex samples. Seven of these miRNAs were experimentally validated, and 19 were replicated in an independent dataset. Some miRNAs showed differential expression between HD and control or PD and control samples, suggesting potential roles in neurodegenerative diseases. These findings underscore the diversity of human miRNAs, especially in tissue-specific contexts, and their potential implications for understanding diseases like HD and PD.
Reference:
- Wake C, Labadorf A, Dumitriu A, et al. Novel microRNA discovery using small RNA sequencing in post-mortem human brain. BMC genomics, 2016, 17(1): 776.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Simultaneous carbon catabolite repression governs sugar and aromatic co-utilization in Pseudomonas putida M2
Journal: Applied and environmental microbiology
Year: 2023
IL-4 drives exhaustion of CD8+ CART cells
Journal: Nature Communications
Year: 2024
High-Fat Diets Fed during Pregnancy Cause Changes to Pancreatic Tissue DNA Methylation and Protein Expression in the Offspring: A Multi-Omics Approach
Journal: International Journal of Molecular Sciences
Year: 2024
KMT2A associates with PHF5A-PHF14-HMG20A-RAI1 subcomplex in pancreatic cancer stem cells and epigenetically regulates their characteristics
Journal: Nature communications
Year: 2023
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch
Journal: Science Advances
Year: 2024
Genomic imprinting-like monoallelic paternal expression determines sex of channel catfish
Journal: Science Advances
Year: 2022
See more articles published by our clients.


