
 Sample Submission Guidelines
Sample Submission Guidelines

What Is Shotgun Metagenomic Sequencing?
Shotgun metagenomic sequencing is a high-throughput, untargeted approach for analyzing the complete genetic content of all microorganisms in a given sample. Unlike amplicon-based methods that rely on specific primers, shotgun sequencing randomly fragments and sequences all extracted DNA — capturing bacterial, viral, fungal, and archaeal genomes in one go.
This method provides a more complete picture of microbiome composition, gene function, and metabolic pathways. It’s especially suited for complex environments like soil, aquatic systems, the human gut, and skin. Researchers also use it to discover novel microbes and rare functional genes not detectable by targeted approaches.

Why Use Shotgun Metagenomic Sequencing?
Shotgun sequencing delivers a comprehensive view of the microbiome — ideal for applications that demand depth, resolution, and functional clarity.

Key Advantages:
- Primer-free detection: Captures full microbial genomes, no prior knowledge required
- High taxonomic resolution: Distinguishes organisms down to the strain level
- Rich functional insights: Identifies genes, pathways, and biological roles
- Wide organism detection: Includes viruses, fungi, and unculturable species
- Supports novel discovery: Enables de novo assembly and gene mining
- Data-rich outputs: Perfect for robustbioinformatics and multi-omics integration
Shotgun vs. Amplicon Sequencing: A Quick Comparison
| Feature | Shotgun Metagenomics | Amplicon Sequencing (e.g., 16S) |
|---|---|---|
| Primer design required | ❌ No | Yes |
| Microbial coverage | All microbes (bacteria, viruses, fungi) | Mostly bacteria and archaea |
| Taxonomic resolution | High (species/strain level) | Limited (genus/species level) |
| Functional gene analysis | Yes | ❌ No |
| Novel species detection | Yes | ❌ Limited |
| Cost & data volume | Higher cost, large datasets | Lower cost, smaller datasets |
| Bioinformatics complexity | High | Low |
Shotgun Metagenomic Sequencing Service Options
CD Genomics offers a range of metagenomic sequencing services tailored to fit diverse research goals and sample types. Depending on your study’s complexity, microbial composition, and desired depth of functional analysis, you can select the most suitable metagenomic sequencing strategy.

Standard Shotgun Metagenomic Sequencing
Broad genome coverage | Taxonomy & function | For varied samples
Learn More ↓
Long-Read Metagenomic Sequencing
Better continuity | Complex regions | Diverse genomes
Explore Long-Read Services →
Sensitive virus detection | Rare viruses | Novel discoveries
Discover Viral Metagenomics Solutions →Shotgun Sequencing Service Workflow

Research goal consultation
Custom protocol design
Sequencing plan confirmation

Sample registration and documentation
DNA quality checks (concentration, purity)
Optional DNA extraction

DNA fragmentation and library prep
Quality validation and method selection

Platforms: Illumina NovaSeq, HiSeq, or DNBSEQ
Read lengths: 2×150 bp or 2×300 bp
Depth customisable based on project goals

Raw sequencing data
Full analysis reports, figures, and summaries
Bioinformatics Services for Shotgun Sequencing
Our in-house bioinformatics team delivers journal-ready reports with clean, high-impact visualisations. From standard QC to advanced ecological statistics, we’ve got you covered.
Core Analysis
- Quality control and read filtering
- Host DNA removal
- Genome assembly and contig stitching
- Gene prediction and clustering (non-redundant)
- Abundance estimation for gene families
- Functional annotation (KEGG, ARDB, CAZyme, eggNOG)
- Taxonomic composition and abundance statistics
- Diversity metrics and community profiling
Advanced Analysis (Optional Add-ons)
- Multi-level taxonomy visualisation
- Group difference analysis (ANOSIM, MRPP)
- Marker species identification (LEfSe)
- Environmental correlation (RDA/CCA)
- Functional significance testing (STAMP: Welch’s t-test, ANOVA, etc.)
- Community diversity analysis (PCA, PCoA, NMDS, clustering)
All results are publication-grade, ready for manuscript inclusion or project reporting.

Applications of Shotgun Metagenomic Sequencing
Unlock comprehensive insights into complex microbial communities with shotgun metagenomic sequencing. At CD Genomics, our platform empowers researchers to explore microbial diversity, identify novel species, and analyze dynamic shifts across diverse environment.
- Microbial Diversity Profiling
Analyze the full spectrum of microorganisms—bacteria, viruses, archaea, fungi—directly from soil, water, or marine samples. This culture-independent approach reveals species-level abundance patterns in natural ecosystems. - Functional Gene and Pathway Discovery
Uncover the ecological roles of microbial communities by detecting genes tied to metabolism and biological processes. Generate a functional map to support environmental monitoring or enzyme discovery. - Novel Microbe and Pathogen Identification
Capture rare, unculturable, or previously undetected microbes using unbiased whole-genome sequencing. This method is vital for bioprospecting and tracing unknown pathogens in environmental or clinical contexts. - Microbiome-Environment Interaction Analysis
Track how microbial populations shift in response to environmental factors, time, or interventions. Frequently used in agriculture, fermentation, and bioremediation research. - Antibiotic Resistance and Mobile Genetic Element Surveillance
Precisely detect resistance genes, plasmids, and transposons. This enables advanced tracking of microbial evolution, gene transfer, and public health threats. - Industrial Strain Screening and Optimisation
Rapidly screen for key enzymes, biosynthesis pathways, or productivity genes to support industrial microbiology, strain engineering, and biomanufacturing.

Sample Requirements for Shotgun Metagenomic Sequencing
| Parameter | Requirement |
|---|---|
| Sample Types | Stool, environmental samples (e.g., soil, water), purified DNA |
| Total DNA | ≥500 ng (minimum 20 ng accepted) |
| DNA Concentration | ≥5 ng/µL |
| DNA Purity (OD260/280) | 1.8–2.0 |
💡Tips:
- Ship samples on dry ice to preserve integrity.
- DNA extraction services available upon request.
- For special sample types or low-input scenarios, contact us for a customised plan.
Why Choose CD Genomics for Your Shotgun Metagenomic Project?
- Strict Quality Control: Sequencing outputs exceed industry benchmarks.
- Platform Flexibility: Choose between short-read and long-read sequencing to fit your study design.
- End-to-End Service: From DNA extraction to library prep and bioinformatics—we manage it all.
- High Throughput Capacity: Capable of processing large sample batches for population-level studies.
- Rapid Turnaround: Automated pipelines ensure fast, consistent data delivery.
- Expert Support: Dedicated project managers provide guidance from planning to interpretation.
- Fully Customisable: Tailor sequencing depth, analysis workflows, and reporting to your research goals.
Whether you're mapping microbial ecosystems or discovering next-generation probiotics, CD Genomics delivers the data clarity and technical precision you need.

Reference
- Joseph, T.A., Pe’er, I. (2021). An Introduction to Whole-Metagenome Shotgun Sequencing Studies. In: Shomron, N. (eds) Deep Sequencing Data Analysis. Methods in Molecular Biology, vol 2243. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-1103-6_6
- Thoendel, Matthew, et al. "Comparison of three commercial tools for metagenomic shotgun sequencing analysis." Journal of clinical microbiology 58.3 (2020): 10-1128. https://doi.org/10.1038/s41598-022-07995-7
- Zuo, Wenxuan, et al. "16S rRNA and metagenomic shotgun sequencing data revealed consistent patterns of gut microbiome signature in pediatric ulcerative colitis." Scientific Reports 12.1 (2022): 6421. https://doi.org/10.1128/jcm.00981-19
- Angeli, Dario, et al. "Insights gained from metagenomic shotgun sequencing of apple fruit epiphytic microbiota." Postharvest Biology and Technology 153 (2019): 96-106. https://doi.org/10.1016/j.envres.2023.116437
- Vijayvargiya, Prakhar, et al. "Application of metagenomic shotgun sequencing to detect vector-borne pathogens in clinical blood samples." PLoS One 14.10 (2019): e0222915. https://doi.org/10.1371/journal.pone.0222915
Demo Results
Partial results are shown below:

Per base sequence conten.
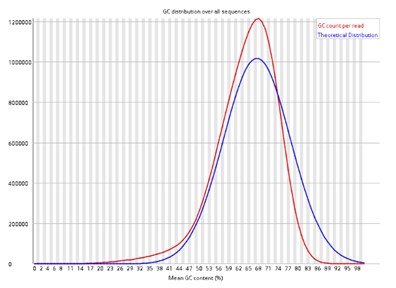
Per sequence GC content.

Merged_abundance.

Abundance heatmap of family level.

KEGG_classification.

CAZy function classification.

Boxplot analysis based on bray Curtis (A), binary jaccard (B), unweighted unifrac (C), and weighted unifrac (D).

PCoA analysis based on bray Curtis (A), binary jaccard (B), unweighted unifrac (C), and weighted unifrac (D).

UPGMA clustering tree.
Metagenomic Shotgun Seq FAQs
1. How can host DNA contamination be minimized in metagenomic projects?
Avoiding host DNA contamination starts at the sampling stage. We recommend collecting material away from host tissues to reduce the inclusion of host cells. Additionally, using host DNA depletion kits during sample preparation can significantly lower contamination.
If a reference genome for the host is available, host-derived sequences can be computationally removed via alignment-based filtering. However, when contamination is severe and no host genome is available, data quality and interpretation may be compromised. In such cases, we recommend resolving contamination issues before sequencing.
2. What are the advantages of metagenomic sequencing over single genome sequencing?
Shotgun metagenomic sequencing captures the structure and functional potential of entire microbial communities, revealing interactions among diverse organisms.
Unlike single-genome sequencing—which targets isolated strains—metagenomics eliminates the need for culturing, making it ideal for exploring complex or unculturable microbial ecosystems. This leads to a more holistic and accurate understanding of microbial ecology.
3. How does 16S rDNA sequencing compare with shotgun metagenomics?
16S rDNA sequencing targets a conserved gene region in bacteria to assess taxonomic composition and diversity. In contrast, shotgun metagenomic sequencing analyzes the entire genomic content of all microorganisms (bacteria, archaea, fungi, viruses), offering both taxonomic and functional insights. While 16S is cost-effective and simpler, shotgun sequencing delivers higher resolution and more comprehensive data—at a higher cost and complexity.
4. How do I choose the appropriate sequencing depth and read length?
Your selection depends on the complexity of the sample and the goals of your study. High-complexity samples (e.g., soil or gut microbiota) require deeper sequencing to capture the full diversity. Common read lengths include 2×150 bp or 2×300 bp. We’ll work closely with you to recommend optimal parameters based on your project’s biological and analytical requirements.
5. How is sequencing data quality ensured?
We utilize advanced high-throughput sequencing platforms such as Illumina NovaSeq and HiSeq, combined with rigorous quality control measures at every stage—from DNA extraction and library preparation to data processing. All datasets undergo multi-step quality filtering to ensure integrity, reliability, and publication readiness.
6. Do you offer custom bioinformatics analysis services?
Yes. We provide fully customizable analysis workflows tailored to your research goals—whether it's functional gene mining, antimicrobial resistance profiling, or advanced statistical modeling. Our experienced bioinformatics team will consult with you throughout the process to ensure the results align with your scientific objectives.
Metagenomic Shotgun Seq Case Studies
Customer Publication Highlight
Abundance and phylogenetic distribution of eight key enzymes of the phosphorus biogeochemical cycle in grassland soils
Journal: Environmental Microbiology Reports
Published: 10 May 2023
DOI: https://doi.org/10.1111/1758-2229.13159
Background
Grassland soils are critical for global phosphorus (P) cycling, with microbial enzymes (e.g., phosphatases, phytases) driving organic P mineralization. This study analyzed 74 global grassland soil metagenomes to map the abundance, diversity, and environmental drivers of eight key P-cycle enzymes, including alkaline phosphatases (phoD, phoX, phoA), acid phosphatases (Nsap-A/B/C), and phytases (BPP, CPhy).
Project Objectives
- Taxonomic & Functional Profiling: Characterize microbial communities and P-enzyme genes.
- Environmental Correlations: Link enzyme abundance to soil pH, SOC, moisture, and climate.
- Strain-Level Resolution: Identify dominant microbial taxa harboring P-enzyme genes.
CD Genomics’ Services
As a key collaborator, CD Genomics provided:
- Shotgun Metagenomic Sequencing
- Platform: Illumina NovaSeq (2×150 bp) for Uruguayan soil samples.
- Coverage: High-depth sequencing (~10 Gb/sample) to capture rare taxa.
- Library Prep:
- DNA shearing (~300–500 bp fragments).
- Dual-indexed libraries for multiplexing.
- Bioinformatics
Pipeline
- Quality Control: FastQC for raw read validation.
- Taxonomic Assignment: Kraken2 + GTDB v214 for species/strain-level resolution.
- Functional Annotation:
- HUMAnN3 (KEGG/MetaCyc pathways).
- CARD/DeepARG for antibiotic resistance genes.
- Phylogenetic Analysis:
- EPA-ng for P-enzyme gene placement.
- Edge-PCA to link enzyme variants to soil types.
- Data Delivery
- FASTQ files + interactive reports (PCoA plots, heatmaps).
- Custom Analysis: Correlation of phoD variants with SOC/clay content.
Key Findings
- Dominant P-Enzymes:
- phoD (alkaline phosphatase) was 5× more abundant than phoX and linked to high SOC/clay soils.
- Acid phosphatases (Nsap-A/C) thrived in acidic soils, while BPP phytases preferred pH > 6.6.
- Environmental Drivers:
- pH and Tmax strongly influenced enzyme distribution (p < 0.001).
- phoD-carrying microbes (e.g., Koribacter, Rhodanobacter) dominated low-pH/high-SOC soils.
- Strain-Level Insights:
- Bacillus and Planctomyces variants correlated with neutral-pH soils.
- Burkholderia phoX genes were enriched in low-SOC soils.
Figures Referenced
 FIGURE 1. Phylogenetic placements of the predicted proteins of each metagenome with respect to the reference bases of each enzyme.
FIGURE 1. Phylogenetic placements of the predicted proteins of each metagenome with respect to the reference bases of each enzyme.
 FIGURE 2. CAP analysis linking phoD abundance to soil pH/SOC
FIGURE 2. CAP analysis linking phoD abundance to soil pH/SOC
 Figure 4. Edge-PCA plots showing phoD variants in Ferralsols vs. Luvisols
Figure 4. Edge-PCA plots showing phoD variants in Ferralsols vs. Luvisols
Implications for CD Genomics Clients
- Agricultural Solutions: Optimize P-cycling microbes for low-input soils.
- Bioremediation: Target phoD-rich taxa for organic P mobilization.
- Custom Services: Strain-level metagenomics + pathway analysis for microbiome studies.
Reference
- Garaycochea, Silvia, et al. "Abundance and phylogenetic distribution of eight key enzymes of the phosphorus biogeochemical cycle in grassland soils." Environmental Microbiology Reports 15.5 (2023): 352-369. https://doi.org/10.1111/1758-2229.13159
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Hydrogen-Oxidizing Bacteria Are Abundant in Desert Soils and Strongly Stimulated by Hydration
Journal: mSystems
Year: 2020
Abundance and phylogenetic distribution of eight key enzymes of the phosphorus biogeochemical cycle in grassland soils
Journal: Environmental Microbiology
Year: 2023
Chitinase Chit62J4 Essential for Chitin Processing by Human Microbiome Bacterium Clostridium paraputrificum J4
Journal: Molecules
Year: 2021
Nutrient structure dynamics and microbial communities at the water–sediment interface in an extremely acidic lake in northern Patagonia
Journal: Frontiers in Microbiology
Year: 2024
Gene functions of the Ambystoma altamirani skin microbiome vary across space and time but potential antifungal genes are widespread and prevalent
Journal: Microbial Genomics
Year: 2024
Shifts in the rhizosphere microbiome and exudation profile of avocado (Persea americana Mill.) during infection by Phytophthora cinnamomi and in presence of a biocontrol bacterial strain
Journal: CABI Agriculture and Bioscience
Year: 2023
See more articles published by our clients.


