
 Sample Submission Guidelines
Sample Submission Guidelines

What Is Bacterial Whole Genome de novo Sequencing
Bacterial whole genome de novo sequencing is a reference-free approach that enables complete reconstruction of bacterial genomes — including both chromosomes and plasmids — directly from sample data.
This technique delivers a full, high-resolution genome map, making it ideal for studying unknown strains, identifying gene functions, and analyzing microbial evolution. It’s especially useful when no reliable reference genome exists or when dealing with genetically complex species.
How It Works: From Sample to Complete Genome
The de novo sequencing process integrates long-read sequencing, high-accuracy short-read correction, and robust assembly tools to ensure precision at every step:
- Long-read sequencing
library preparation
Platforms like PacBio HiFi or Oxford Nanopore generate continuous reads of 10–25 kb or longer. These long reads span repetitive and structurally complex regions that are difficult to resolve using short-read data alone. - Reference-free genome assembly
Assembly tools such as Hifiasm and Canu stitch together these long reads into full genomes — entirely from scratch, without relying on existing reference sequences. - Short-read polishing
High-accuracy data from Illumina sequencing is layered on to correct minor errors, improving the reliability and base-level accuracy of the assembly. - Multi-step genome annotation
Final quality control and functional annotation pipelines ensure that your data is not only complete, but biologically meaningful — ready for downstream analysis.
 Whole Genome de novo Sequencing process
Whole Genome de novo Sequencing process
Why Bacterial Whole Genome de novo Sequencing Is Essential for Your Research

Designed specifically for bacterial samples lacking a reference genome or with limited reference data, de novo whole genome sequencing enables accurate, complete genome assembly. This approach reveals complex genetic variations and repetitive regions, significantly improving assembly quality and accuracy.
- Accurate, Complete Genome Assembly
Assemble chromosomes and plasmids with high continuity—without relying on any reference sequences. - Comprehensive Genome Analysis
Detect structural variations, functional genes, antimicrobial resistance markers, and repetitive elements for a full genetic profile. - Integration of Advanced Sequencing Technologies
Combine long-read sequencing with high-throughput short reads to ensure data accuracy and depth. - Versatile for Diverse Strains
Suitable for novel, complex, or hard-to-sequence bacterial strains, guaranteeing reliable assembly outcomes.
Our Whole Genome de novo Sequencing Portfolio: Tailored for Bacteria, Fungi, and More
CD Genomics offers species-specific de novo genome sequencing services to meet diverse research needs without the limitations of a reference genome.

Bacterial Whole Genome de novo Sequencing
Reference-free assembly | Complete genome reconstruction | Structural variation discovery
View Bacterial WGS Details ↓
Fungal Whole Genome de novo Sequencing
High-contiguity assembly | Repeat-rich genome resolution | Functional annotation ready
Explore Fungal WGS Service →
De Novo Whole Genome Sequencing Service
Multi-species support | Long-read + short-read integration | Ideal for novel species
Learn More About Multi-species de novo WGS →Streamlined Workflow for Bacterial Whole Genome de novo Sequencing: From Sample to Insights

≥10 µg high-quality DNA
OD260/280 = 1.8–2.0

PacBio / Nanopore / Illumina
Long and short insert libraries

de novo assembly tools
Multi-round polishing
Hybrid error correction

Gene prediction and annotation
Resistance/virulence gene identification
Functional and comparative genomics

Quality control metrics
Visual reports and data summary
Optimized Sequencing Strategies for Bacterial Whole Genome de novo Assembly
Library Construction Highlights:
- Multiple insert size libraries designed for optimal coverage, including short inserts (~350 bp) and long inserts (5–20 kb).
- PCR-free library preparation to minimize amplification bias.
- Strict quality control to ensure even data coverage.
Sequencing Platforms:
- PacBio Sequel IIe / Revio: Produces highly accurate HiFi long reads (10–25 kb) with >Q20 accuracy, ideal for assembling complex genomes continuously.
- Oxford Nanopore PromethION: Offers ultra-long reads reaching megabase scale, enhancing assembly of highly complex regions.
- Illumina NovaSeq 6000: 150 bp paired-end sequencing with deep coverage and >90% bases at Q30 quality, perfect for error correction and precision polishing.
Recommended Sequencing Depth:
- Standard: >100× coverage with PacBio HiFi reads.
- Supplementary: >50× coverage with Illumina short reads to ensure high-accuracy genome correction.
Data Quality Metrics:
- HiFi read accuracy above 99.9%.
- Illumina data with over 90% bases at Q30 quality or higher.
- High data integrity with excellent assembly continuity.
Advanced Bioinformatics Analysis: Turn Bacterial Genome Data into Actionable Insights
We offer professional, efficient, and comprehensive bioinformatics analysis services to unlock the full potential of your bacterial genome data and accelerate research progress.
Standard Analysis – Ensuring Data Quality and Accuracy
- Data Quality Control and Cleaning: Remove low-quality and contaminant sequences to ensure reliable downstream analysis.
- Genome Assembly and Scaffolding: Use advanced algorithms to produce continuous, complete bacterial genome sequences.
- Sequence Correction: Perform multiple rounds of error correction to reduce sequencing errors and improve annotation quality.
- Gene Prediction: Accurately identify protein-coding genes and non-coding RNAs for a thorough understanding of genome function.
- Functional Annotation: Integrate databases like GO, KEGG, and eggNOG to define gene biological roles and metabolic pathways.
- Repeat Sequences and CRISPR Prediction: Detect complex genome structures and bacterial defense systems for in-depth functional insights.
Advanced In-Depth Analysis – Extracting Additional Biological Insights
- Virus and Phage Prediction: Identify potential prophage sequences within bacterial genomes to reveal microbial interactions.
- Virulence Factors and Resistance Gene Analysis: Precisely detect virulence and antibiotic resistance genes to support pathogen studies and resistance monitoring.
- Carbohydrate-Active Enzymes (CAZy) Analysis: Explore genes coding for metabolic enzymes, aiding industrial enzyme and metabolism research.
- Transmembrane Proteins and Signal Peptide Prediction: Predict key membrane proteins and signal peptides to assist drug target discovery and functional studies.
- Comparative Genomics: Conduct phylogenetic trees, gene family clustering, and synteny analysis to study strain evolution and functional differences.

Sample Requirements for Bacterial Whole Genome de novo Sequencing
| Sample Type | Requirement Description |
|---|---|
| Total DNA Amount | ≥ 10 μg |
| DNA Concentration | ≥ 80 ng/µL |
| DNA Purity | OD260/OD280 ratio between 1.8 and 2.0 |
| Integrity | No visible degradation or RNA contamination; verified intact by gel electrophoresis |
Sample Submission Recommendations:
- Use low-binding centrifuge tubes free of DNases, such as 1.5 mL Eppendorf tubes, to store samples.
- For short-term transport, keep samples chilled with ice packs; for longer transport, use dry ice.
- Clearly label each sample with an identifiable number.
Applications of Bacterial Whole Genome de novo Sequencing
Our Bacterial Whole Genome de novo Sequencing service provides a comprehensive view of bacterial genomes, supporting a wide range of research areas:

- Antibiotic Resistance Mechanisms
Precisely locate resistance gene islands such as β-lactamases, aiding in understanding drug resistance and guiding public health interventions. - Tracking Virulence Evolution
Analyze the horizontal transfer of virulence factors to support studies on pathogen evolution and virulence changes. - Industrial Strain Optimization
Identify key metabolic pathway genes to enhance fermentation efficiency and improve bacterial strains. - Environmental Adaptation Mechanisms
Reveal survival strategies of bacteria in extreme environments, benefiting ecological and environmental research. - New Species Identification
Construct complete genomic profiles to facilitate microbial taxonomy and the discovery of novel species.
Why Choose CD Genomics for Bacterial Whole Genome de novo Sequencing?
CD Genomics delivers a trusted, one-stop service offering high-quality, fast turnaround, and comprehensive analysis for bacterial whole genome de novo sequencing. Our focus extends beyond sequencing to ensuring top-notch data quality and biologically meaningful results.
- Multi-Platform Integrated Sequencing Strategy
Combine the strengths of PacBio HiFi, Oxford Nanopore, and Illumina platforms to achieve high accuracy and complete genome assemblies. - Customized Assembly and Correction Pipelines
Tailor assembly protocols based on sample characteristics, applying multiple rounds of error correction using third-generation raw data, self-alignment, and second-generation data polishing to ensure highly accurate final sequences. - Comprehensive Bioinformatics Analyses
From raw data processing to functional annotation, phylogenetics, resistance genes, virulence factors, and metabolic pathways, our analyses support diverse research goals. - Rigorous Quality Control System
Follow standardized protocols from sample receipt through library preparation, sequencing, and assembly QC, guaranteeing consistent and reliable data delivery. - Clear, Visual, and Actionable Data Reports
Provide easy-to-interpret charts and structured annotation files, facilitating downstream analyses and manuscript preparation. - Dedicated Technical Support
Receive expert guidance throughout your project, including experimental design, data interpretation, and troubleshooting, helping you navigate complex analysis challenges smoothly.

Demo Results
Partial results are shown below:

Distribution of base quality

Distribution of base content

Shared SNP number between samples

SNP mutation type distribution
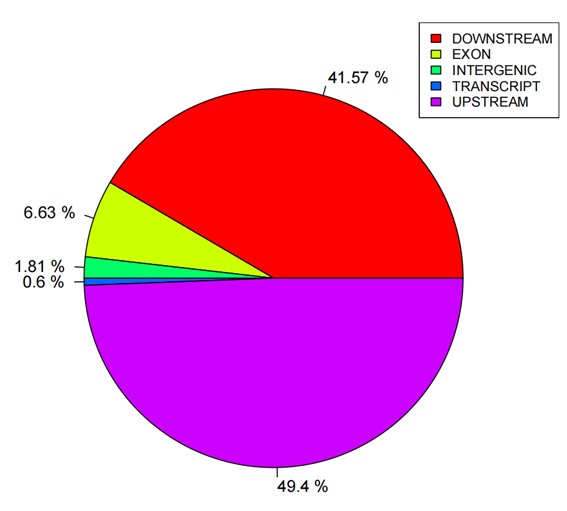
Statistics pie of SNP annotations

InDel length distribution
Bacterial Whole Genome de novo Seq FAQs
1. What indicators can be used to evaluate bacterial genome assembly?
The common indicators for the quality of genome assembly include scaffold N50, N%, scaffold numbers, and the total number of base pairs.
2. How to achieve zero gap?
Currently, the complete sequence map of more than 90% bacterial strains can be constructed by making use of a combination of Illumina HiSeq and PacBio SMRT systems. Pacbio RS II system can achieve complete genome assembly even in the regions of high or low GC content, as well as repetitive sequences. The complete sequence map of the rest 10% bacterial strains can be achieved with Sanger sequencing data. CD Genomics has completed hundreds of bacterial genome assembly cases without gap.
3. Is it feasible to complete a bacterial genome using only third-generation single-molecule sequencing platforms?
No, it is not feasible. Small plasmid fragments (approximately 20 kb) may be lost during the library construction process. Additionally, certain regions of the chromosome may not be sequenced due to sampling probability issues or sample degradation.
4. How can we ensure the accuracy of the assembly given the low single-base accuracy of third-generation single-molecule sequencing platforms?
The single-base accuracy of third-generation single-molecule sequencing data ranges between 87% and 92%. To ensure the accuracy of the assembly, we can employ the following three-step process:
- Prior to assembly, correct the sequencing data by leveraging the overlap between third-generation single-molecule sequencing sequences.
- Post-assembly, use third-generation single-molecule sequencing data to correct the assembled sequences.
- After the second correction, use high-quality second-generation high-throughput sequencing data for further correction of the assembled results.
By applying this three-step correction process, the final assembly accuracy can exceed 99.99%.
5. How does long-read sequencing address repetitive regions in bacterial genomes?
The 15-25kb extended read lengths offer a unique solution:
- They effectively span and fully cover repetitive units, such as IS elements and rRNA clusters.
- Avoid assembly breaks commonly caused by short-read sequencing.
- Demonstrated over 99% assembly completeness in repetitive sequence areas.
6. Is a separate experiment required for epigenetic detection (6mA/4mC)?
No, there’s no need for additional experiments. With PacBio HiFi technology:
- Base modifications are captured natively without extra library preparation or sequencing efforts.
- It directly provides a comprehensive whole-genome methylation map.
- Sensitivity: Detects sites with a modification frequency of ≥85% with over 95% accuracy.
7. Does abnormal GC content (<20% or >80%) affect results?
PacBio technology neutralizes GC bias:
- Ensures coverage differences across 15-85% GC regions are under 5%.
- No need for specialized library optimization.
Bacterial Whole Genome de novo Seq Case Studies
Customer Publication Highlight
Phenotypic and Draft Genome Sequence Analyses of a Paenibacillus sp. Isolated from the Gastrointestinal Tract of a North American Gray Wolf (Canis lupus)
Journal: Applied Microbiology
Impact Factor: ~4.5 (2023)
Published: 23 September 2023
DOI: https://doi.org/10.3390/applmicrobiol3040077
Background
Canine inflammatory bowel disease (cIBD) lacks effective treatments, with gut dysbiosis as a key factor. Gray wolves (Canis lupus), ancestors of domestic dogs, harbor unique gut microbiota potentially lost during domestication. This study isolated a spore-forming Paenibacillus sp. strain from a wild wolf GI tract, characterizing its probiotic potential for cIBD treatment.
Project Objectives
- Isolate & Phenotype: Recover chloroform-resistant spore-formers from wolf GI tract; assess antimicrobial activity.
- Genomic Analysis: Sequence and annotate the genome to identify probiotic-associated genes.
- Phylogenetic Typing: Determine taxonomic identity and evolutionary relationships.
CD Genomics’ Services
As the genomics partner, CD Genomics delivered:
- Whole Genome Sequencing (WGS)
- Platform: Illumina NovaSeq (400 Mbp reads).
- Coverage: Draft assembly (7,034,206 bp).
- Library Prep: DNA extraction.
- Bioinformatics Analysis
- Assembly & Annotation: JGI IMG/MER pipeline for gene prediction (6,543 genes).
- Functional Annotation: COG categorization, conserved domain analysis (CD-Search).
- Prophage Detection: PHAge Search Tool (PHASTER) for lysogenic sequences.
- Phylogenetics: BLAST+/MEGA for 16S rRNA typing; Mugsy/RAxML for whole-genome phylogeny.
Key Findings
- Probiotic Phenotype Validated
- Antimicrobial Activity: Inhibited Staphylococcus aureus, Escherichia coli, and Micrococcus luteus (Figure 1B, Supplementary Fig S1).
- Enzyme Production: Starch hydrolysis (Figure 1A), lipase, and cellulase activity confirmed.
- Safety Profile: Antibiotic-sensitive (tetracycline, erythromycin); no toxin genes detected.
- Genomic Insights via CD Genomics’ WGS
- Antimicrobial Genes: Bacteriocins (5), lantibiotics (6), chitinases (2), lysins (22), amidases (42) (Table 1).
- Metabolic Enzymes: Alpha-amylase, cellulase, lipases, pectin lyase—critical for carbohydrate digestion.
- Sporulation: 133 genes for spore formation/germination (enhancing probiotic survivability).
- Viral Elements: 48 phage-derived genes (non-functional, antimicrobial potential).
- Taxonomic Classification
- 16S rRNA Typing: 99% identity to Paenibacillus xylanexedens PAMC 22703.
- Phylogenomics: Closest relatives: P. amylolyticus SQR-21 (drought-resistant wheat associate) and Paenibacillus sp. OVF10 (medicinal plant isolate) (Figure 3).
Figures Referenced
 Figure 2. Conserved domain analysis: (A) Outer spore coat, (B) Sporulation protein K, (C) Penicillin-binding, (D) Antibiotic synthesis.
Figure 2. Conserved domain analysis: (A) Outer spore coat, (B) Sporulation protein K, (C) Penicillin-binding, (D) Antibiotic synthesis.
 Figure 3. Phylogenetic tree of ClWae2A and related Paenibacillus spp. (Bootstrap >83%).
Figure 3. Phylogenetic tree of ClWae2A and related Paenibacillus spp. (Bootstrap >83%).
Implications
- Probiotic Development: ClWae2A’s spore formation, pathogen inhibition, and carbohydrate-digesting enzymes position it as a candidate for canine IBD treatment.
- Microbiome Restoration: Reintroducing wolf-derived bacteria may counter dysbiosis caused by domestication.
- Precision Genomics: CD Genomics’ WGS enabled identification of safety markers (no toxins) and functional genes (antimicrobials/enzymes), de-risking probiotic design.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Identification of diverse integron and plasmid structures carrying a novel carbapenemase among Pseudomonas species
Journal: Front. Microbiol.
Year: 2019
Production of a Bacteriocin Like Protein PEG 446 from Clostridium tyrobutyricum NRRL B-67062
Journal: Probiotics and Antimicrobial Proteins
Year: 2024
Untangling the Role of Pathobionts from Bacteroides Species in Inflammatory Bowel Diseases
Journal: bioRxiv
Year: 2023
A chromosome-level genome resource for studying virulence mechanisms and evolution of the coffee rust pathogen Hemileia vastatrix
Journal: bioRxiv
Year: 2022
Streptomyces buecherae sp. nov., an actinomycete isolated from multiple bat species
Journal: Antonie Van Leeuwenhoek
Year: 2020
See more articles published by our clients.


