
 Sample Submission Guidelines
Sample Submission Guidelines
Dual RNA-seq
Dual RNA-seq has been shown to monitor the all classes of coding and noncoding transcripts of both host and pathogen simultaneously. CD Genomics is offering a high-resolution, affordable and straightforward dual RNA-seq service to provide direct insight into host–pathogen interplay.
What Is Dual RNA-seq
There is a wide range of interactions between species, such as parasitism, symbiosis, competition, etc. The conventional transcriptome sequencing can only study the information of a single species, which not only wastes part of the data, but also affects the sample itself during the separation of two species.
The sequencing technology Dual RNA-seq was developed precisely to address these types of inquiries. By constructing only one transcriptome library, dual RNA-seq of total mixed RNA following double rRNA depletion or poly(A) capture allows sequencing and analyzing two (or more) species at the same time without the need to separate the species, thereby revealing the dynamic changes in gene expression between them. Meanwhile, through the interaction model diagram, to obtain the regulatory relationship of genes and the interaction mechanism between two species; to investigate the regulatory network in the interaction process, the mechanism of pathogen infection and the host resistance to disease; and to investigate the evolutionary relationship of pathogen among different species, and further explore the positive selection of related genes based on homologous genes.
CD Genomics could deal with various invasion models-- pathogens involving bacteria, fungi, protozoa, etc., host could be a mammal or plant. We are aiming to provide comprehensive dual RNA-seq services from experimental design to biocomputational analysis to support your research needs.
Advantages of Our Dual RNA-seq Service
- Available for various invasion models
- Flexibility of sample type: total mixed RNA, infected host cells, etc.
- Molecular barcodes technology allows small amounts of input template
- Comprehensive analysis, high stability: Utilizing advanced Illumina platform sequencing, combined with High Performance Computing (HPC), to achieve rapid and stable sequencing data analysis and delivery.
- Strict quality control, diverse analysis pipelines: Implementing rigorous quality control measures and offering diverse analysis pipelines. Not only providing transcriptome analysis for species, but also configuring advanced analyses such as Weighted Gene Co-expression Network Analysis (WGCNA), virulence factor annotation, protein-protein interaction network analysis, etc., to deeply explore inter-species relationship information.
- Scientific and meticulous design: From sample selection, library preparation, sequencing, to data analysis, each step undergoes scientific and meticulous design to ensure high-quality research outcomes.
Applications of Dual RNA-seq
- Plant disease and resistance-susceptibility mechanisms.
- Physiological adaptation and molecular responses.
- Pathogens and host immunity.
- Interspecies symbiosis and evolutionary mechanisms.
- Parasitism in pest and host defense.
- Co-cultivation of bacteria/fungi.
- Infectious disease research.
- Comparative genomics and pathogenic mechanisms.
Workflow of Dual RNA-seq
Upon preparation of your sample, CD Genomics offers a comprehensive suite of sequencing and bioinformatics analysis services.

Service Specification
Sample Requirements:
|
|
 |
Sequencing:
|
 |
Data Analysis
|
Analysis Pipeline
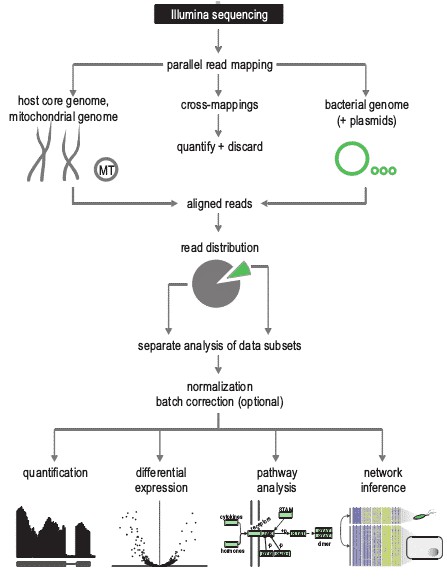 Generic Pipeline of Dual RNA-seq Data Analysis (figure from V. Arluison et al., 2018)
Generic Pipeline of Dual RNA-seq Data Analysis (figure from V. Arluison et al., 2018)
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Dual RNA-seq for your writing (customization)
Dual RNA-sequencing (Dual RNA-seq) is a state-of-the-art sequencing technique developed to address such challenges. Specializing in the investigation of host-pathogen interactions, CD Genomics utilizes an advanced dual RNA-seq data analysis to propel scientific advancements in this field. Our customized solutions, bioinformatics expertise, and unwavering commitment to quality, ensure researchers gain accurate, reliable, and meaningful insights into the intricate world of host-pathogen interactions. For more information about our services, we invite you to get in touch with us.
References:
- Alexander J. Westermann, et al., Dual RNA-seq unveils noncoding RNA functions in host–pathogen interactions. Nature. 2016, vol. 000.
- Alexander J. Westermann, et al., Resolving host–pathogen interactions by dual RNA-seq. PLoS Pathog. 2017, 13(2).
- Véronique Arluison and Claudio Valverde (eds.), Bacterial Regulatory RNA: Methods and Protocols. Methods in Molecular Biology. 2018, vol. 1737.
- Pisu et al., Dual RNA-seq of Mtb-infected macrophages in vivo reveals ontologically distinct host-pathogen interactions. Cell Reports. 2020, vol. 30.
- Nuss A M, Beckstette M, Pimenova M, et al. Tissue dual RNA-seq allows fast discovery of infection-specific functions and riboregulators shaping host–pathogen transcriptomes. Proceedings of the National Academy of Sciences, 2017, 114(5): E791-E800.
Demo Results
Partial results are shown below:

Sequencing quality distribution

A/T/G/C Distribution

IGV Browser Interface

Correlation Analysis Between Samples

PCA Score Plot

Venn Diagram

Volcano Plot

Statistics Results of GO Annotation

KEGG Classification
Dual RNA-seq FAQs
1. Are reference genomes required for all species undergoing Dual RNA-seq?
Ideally, the availability of reference genomes for both interacting species would facilitate more accurate results. However, literature also documents procedures where only a single species has a reference genome at disposal. The analysis workflow in such cases entails initially mapping the sequencing data to the species with a reference genome. The transcriptomic data, post exclusion of mapping data, can be assayed for the other species' information through mapping with a close relative or a de novo assembly, facilitating subsequent analyses. Specifically, for the prokaryotic segment in an interacting sample, the presence of a reference genome is imperative. However, in its absence, bacterial 'pan-genome' profiling can be implemented.
2. What are the advantages of interacting transcriptomes over ordinary transcriptomes?
Dual RNA-seq is a specialized sequencing technique designed to probe interspecies interactions, generally encompassing symbiotic, parasitic, competitive, and predatory relationships. Traditional transcriptomics necessitate the separation of these interacting species, an approach which, albeit useful, can lead to loss of pertinent information and introduce potential artifacts related to the segregation process. In stark contrast is the dual RNA-seq approach, which enables simultaneous investigations of two or multiplex interacting species without the need for their partitioning. Consequently, this circumvents the possible detrimental influence incurred due to species separation. Moreover, this methodology is cost-effective, necessitating the construction of only one transcriptome library, which enables concurrent analysis of all included species.
3. What are the different types of RNA-seq?
Furthermore, depending on the research objectives and peculiarities of the sample, RNA-seq can be subdivided into several types such as: Total RNA-seq, mRNA-seq, miRNA-seq, Long Non-coding RNA-seq, Full-Length Transcriptome Sequencing, Circular RNA-seq, and Whole Exome RNA-seq.
Dual RNA-seq Case Studies
Dual RNA-Seq Uncovers Metabolic Amino Acids Dependency of the Intracellular Bacterium Piscirickettsia salmonis Infecting Atlantic Salmon
Journal: Frontiers in microbiology
Impact factor: 6.064
Published: 27 November 2018
Abstract
High-throughput sequencing technologies, applied to transcriptomic research (RNA-Seq), have opened up the possibility of comprehending complex molecular reactions within different biological contexts. Recently, the synchronous sequencing of a pathogen's and host's transcriptomes during infection - known as dual RNA-seq - has emerged as a promising method to unveil the intricacies of host-pathogen interactions. In this particular study, through dual rRNA depletion and RNA sequencing scheme, researchers simultaneously analyzed the transcriptomes of the intracellular bacterium Piscirickettsia in salmon and its host species, Salmo salar, throughout the course of the infection.
Methods
- Head kidney and spleen tissues
- Total RNA Isolation
- Remove both bacterial and host rRNAs
- Dual RNA-Seq
- Illumina HiSeq platform
- 100 bp paired-end reads
- Differential expression analysis
- Functional annotation
- Gene Ontology (GO) annotation
- KEGG pathway annotation analysis
- qPCR validations
Results
Exploring Host and Pathogen Transcriptome During Pathogenesis
Dual RNA-Seq analysis delineated the regulation of the transcriptomes of P. salmonis and S. salar during the course of infection. Regarding the S. salar transcriptome, 771 and 829 genes were differentially expressed in the spleen at 7 and 14 dpi respectively (Fig 1B). Concurrently, 412 and 467 genes were differentially regulated at these same time points in the head kidney. Genes specifically modulated in the head kidney included those encoding MATE efflux family protein family protein, DNA repair protein RecN, chaperone protein HtpG and various membrane proteins (Fig 2). Among the shared genes, different protein phosphatase 1, ribosomal proteins, molecular chaperones and Asn/Gln transamidosome subunits were identified (Fig 2).
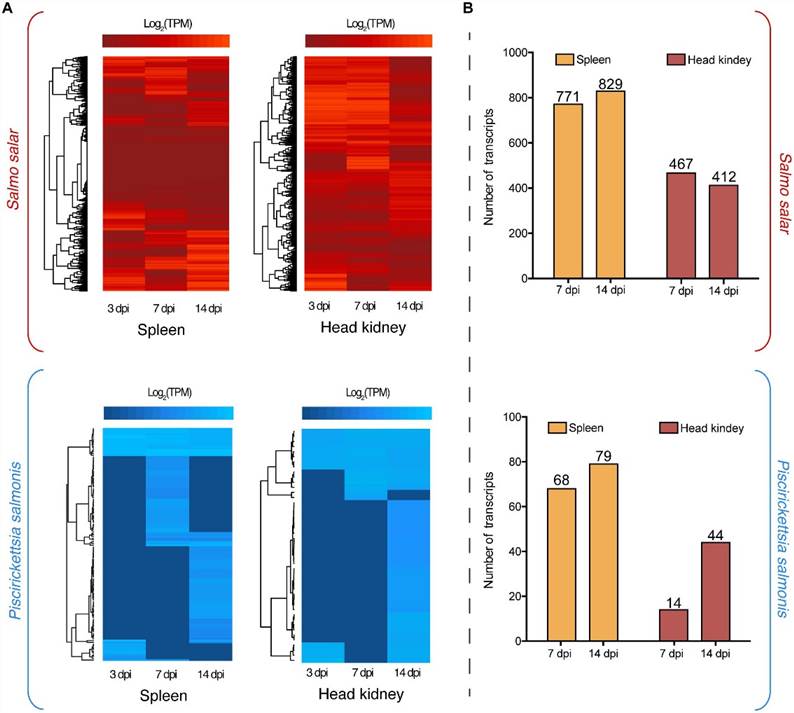 FIGURE 1. (A) Global transcriptome analysis of Piscirickettsia salmonis (blue) and S. salar (red) at 3, 7, and 14 days post-infection (dpi) in spleen and head kidney tissues. Expression values were estimated as transcripts per million reads (TPM) and Log2 transformed for heat map clustering. (B) Differentially expressed genes in spleen and head kidney at 7 and 14 dpi using as control the expression values at 3 dpi. Three days post-infection were used as baseline gene expression since no bacterial reads would be present in any type of control group.
FIGURE 1. (A) Global transcriptome analysis of Piscirickettsia salmonis (blue) and S. salar (red) at 3, 7, and 14 days post-infection (dpi) in spleen and head kidney tissues. Expression values were estimated as transcripts per million reads (TPM) and Log2 transformed for heat map clustering. (B) Differentially expressed genes in spleen and head kidney at 7 and 14 dpi using as control the expression values at 3 dpi. Three days post-infection were used as baseline gene expression since no bacterial reads would be present in any type of control group.
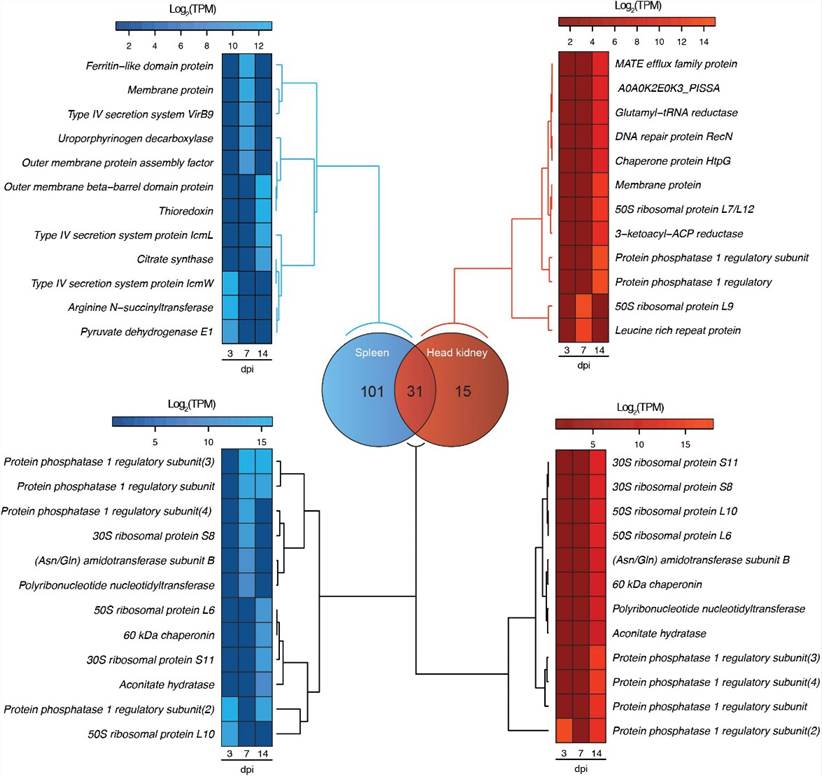 FIGURE 2. Venn diagram showing the number of exclusive and shared genes in the transcriptome of P. salmonis during the infection in spleen (blue) and head kidney (red) tissues of Atlantic salar. The heat maps show a subset of exclusive and shared genes expressed by P. salmonis in both tissues.
FIGURE 2. Venn diagram showing the number of exclusive and shared genes in the transcriptome of P. salmonis during the infection in spleen (blue) and head kidney (red) tissues of Atlantic salar. The heat maps show a subset of exclusive and shared genes expressed by P. salmonis in both tissues.
Amino Acid Metabolism: A Common Response Between P. salmonis and S. salar
The GO enrichment analysis indicated that a large proportion of the differentially expressed genes in P. salmonis were associated with protein and nitrogen compound metabolic processes. Enrichment of genes related to protein metabolism was also discovered via KEGG annotation. However, despite amino acid metabolism not being the major transcriptomic response within the host, both P. salmonis and S. salar demonstrated numerous genes connected to the biological synthesis and degradation of amino acids.
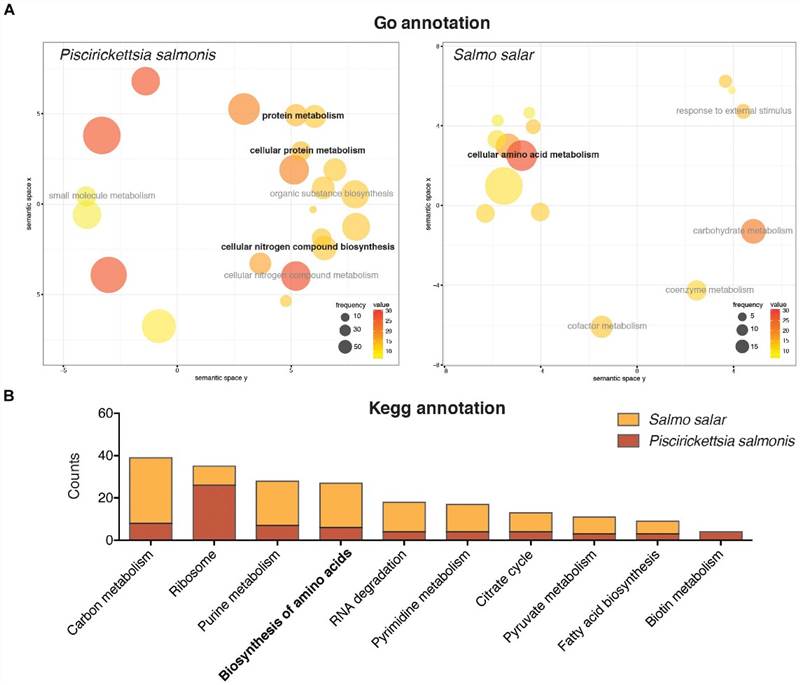 FIGURE 3. Gene ontology annotation (A) and KEGG pathway annotation (B) of the differentially expressed genes in Salmo salar and P. salmonis during the infection.
FIGURE 3. Gene ontology annotation (A) and KEGG pathway annotation (B) of the differentially expressed genes in Salmo salar and P. salmonis during the infection.
Conclusion
Beyond the standard host immune responses and the virulence factors of pathogens, both bacteria and host exhibit a profusion of genes associated with metabolic processes, in particular, those relating to amino acid metabolism. Our findings point to a dependency of P. salmonis on the amino acid metabolism of S. salar, which could suggest a novel pathogenesis mechanism grounded in the capacity to absorb nutrients from the host. In broad terms, dual transcriptome sequencing permits a varied perspective on the interaction between host and pathogen, transcending beyond the biological processes intertwined with immunity.
Reference:
- Valenzuela-Miranda D, Gallardo-Escárate C. Dual RNA-Seq uncovers metabolic amino acids dependency of the intracellular bacterium Piscirickettsia salmonis infecting Atlantic salmon. Frontiers in microbiology, 2018, 9: 418416.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Chaperone-Mediated Autophagy Controls Proteomic and Transcriptomic Pathways to Maintain Glioma Stem Cell Activity
Journal: Cancer research
Year: 2022
Circular DNA tumor viruses make circular RNAs
Journal: Proceedings of the National Academy of Sciences
Year: 2018
Repeated immunization with ATRA-containing liposomal adjuvant transdifferentiates Th17 cells to a Tr1-like phenotype
Journal: Journal of Autoimmunity
Year: 2024
Role of the histone variant H2A.Z.1 in memory, transcription, and alternative splicing is mediated by lysine modification
Journal: Neuropsychopharmacology
Year: 2024
FAK loss reduces BRAFV600E-induced ERK phosphorylation to promote intestinal stemness and cecal tumor formation
Journal: Elife
Year: 2023
Identification of circular RNAs regulating cardiomyocyte proliferation in neonatal pig hearts
Journal: JCI insight
Year: 2024
See more articles published by our clients.


