
 Sample Submission Guidelines
Sample Submission Guidelines
Degradome Sequencing
CD Genomics is now able to provide the degradome sequencing service to facilitate a more comprehensive insight into plant microRNA landscape. By using our service, you can detect the mRNA targets of the microRNA in a highly sensitive and accurate manner.
The Introduction of Degradome Sequencing
MicroRNAs (miRNAs) are a class of endogenous non-coding RNAs of 20-24 nucleotides (nt) in length, produced by highly precise excision from stem-loop precursors. MicroRNAs are important regulators of gene expression at the transcriptional and post-transcriptional levels. The mature miRNA is recruited into an RNA-induced silencing complex (RISC) to degrade mRNA targets and suppress their translation. miRNAs are highly conserved among species based on comparisons among different species. There are also non-conserved and species-specific miRNAs in plants. Non-conserved miRNAs are often expressed at low levels, and, therefore, many are not identified in small-scale sequencing projects. Small RNA sequencing technology has allowed the identification of low-abundance miRNAs.
Modified 5' RACE (rapid amplification of cDNA ends) was widely used for target confirmation and cleavage site mapping. Nevertheless, this approach is laborious, time-consuming, costly, and only applicable to fine-scale investigations. Recently, Degradome sequencing, also referred to as parallel analysis of RNA ends (PARE), has emerged as a powerful method that combines both high-throughput sequencing and modified 5' RACE to screen for miRNA and ta-siRNA (trans-acting siRNA) targets in a large scale. In this method, degraded capped mRNA is adapter-ligated and reverse-transcribed. Fragments are then Mmel-digested, purified, 3'-adapter-ligated, and PCR-amplified. Deep sequencing of the cDNA and degradome analysis provide comprehensive information about uncapped transcripts that undergo degradation in plants.
Advantages of Degradome Sequencing
- Identification of known and novel miRNAs and ta-siRNA
- Identification of miRNA targets and regulatory networks
- Statistical summary of mRNA degradation sites
- Explores novel biomarkers and circRNAs regulatory networks
- High-throughput and high-resolution
Degradome Sequencing Workflow
The general workflow for degradome sequencing is outlined below. To construct degradome sequencing library, the first step is to ligate polyA-RNA samples to RNA adaptor containing a 3'Mme I site, and transcribed. After second-strand synthesis, Mme I digestion, gel purification, and PCR amplification, 3' adaptor is ligated for deep sequencing. Our highly experienced expert team executes quality management, following every procedure to ensure confident and unbiased results.

Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
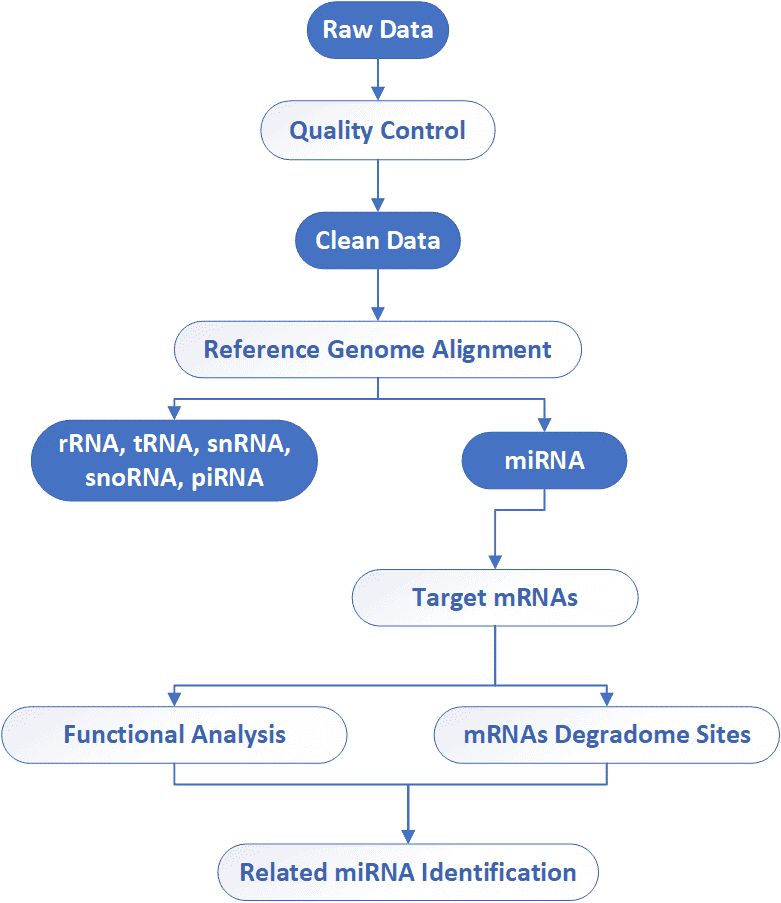
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Degradome Sequencing for your writing (customization)
Supported by our experienced scientists and advanced technology, CD genomics can help you identify small RNA targets at single-base resolution at one time through the high-throughput sequencing by strict quality control and advanced bioinformatics analyses. If you have additional requirements or questions, please feel free to contact us.
Demo Results
Partial results are shown below:

Sequencing quality distribution

A/T/G/C Distribution

IGV Browser Interface

Correlation Analysis Between Samples

PCA Score Plot

Venn Diagram

Volcano Plot

Statistics Results of GO Annotation

KEGG Classification
Degradome Seq FAQs
1. What is the principle of degradome sequencing?
In animals, protein repression is believed to occur by translational inhibition along with mRNA degradation. In plants, miRNAs degrade their mRNA targets by precise cleavage between the 10th and 11th nucleotide s from the 5' end of the miRNA in the complementary region of the target transcript, generating a distinct peak of degradome sequence tag at the predicted cleavage site relative to other regions of the transcript. Based on this principal, the degradome sequencing is primarily used to globally identify remnants of small RNA-directed cleavage by sequencing the 5' ends of uncapped RNAs in plants.
2. What are the advantages of degradome sequencing for identification of miRNA targets?
Table 1. The comparison of degradome sequencing and traditional methods for miRNA target prediction.
| Degradome sequencing | Luciferase reporter gene assay | Argonaute-RNA Immunoprecipitation (AGO-RIP) | 5' RACE (rapid amplification of cDNA ends) | |
| Throughput | High | Low | Low | Low |
| operation | Easy | Complicated | Complicated | Easy |
| period | Short | Long | Long | Long |
| accuracy | High | High | Relatively high | Relatively high |
3. How is degradome sequencing different from other RNA sequencing methods?
Degradome sequencing specifically focuses on RNA degradation products, while other RNA sequencing methods like RNA-Seq typically analyze the entire transcriptome. This makes degradome sequencing uniquely suited for studying miRNA/siRNA-mediated RNA cleavage events.
Reference
- Riffo-Campos, Á.L., et al. Tools for sequence-based miRNA target prediction: What to choose?. International journal of molecular sciences, 2016, 17(12): 1987.
Degradome Seq Case Studies
Small RNA and degradome sequencing reveals important microRNA function in Astragalus chrysochlorus response to selenium stimuli
Journal: Plant biotechnology journal
Impact factor: 6.305
Published: 21 May 2015
Abstract
Astrgalus species are known as hyperaccumulator of Se by converting it to nonaminoacid compounds. But we have no idea about the Se-metabolism-related hyperaccumulation. The authors attempted to understand whether miRNAs play a role in Se accumulation in plants. In this study, they identified 418 known miRNAs and 151 novel miRNAs induced by Se exposure in Astragalus chrysochlorus. Through deep degradome sequencing the authors revealed important miRNA function in A. chrysochlorus response to selenium stimuli.
Materials & Methods
Samples
Callus tissues of A. chrysochlorus seeds;
Selenium treatment.
Sequencing
RNA isolation
RNA quality and quantity measurements
Small RNA sequencing
Degradome sequencing
miRNA identification
Target prediction
Function classification based on GO and KEGG analyses
Results
1. miRNA identification and expression profiles.
A total of 418 known and 151 novel miRNAs was identified. The distribution of miRNAs between control and Se treatment is depicted in figure 1. The 418 miRNAs belong to 380 miRNA families, and 160 miRNA families were differently expressed in both control and Se-treated samples. 30 novel miRNAs were differently expressed after Se treatment.
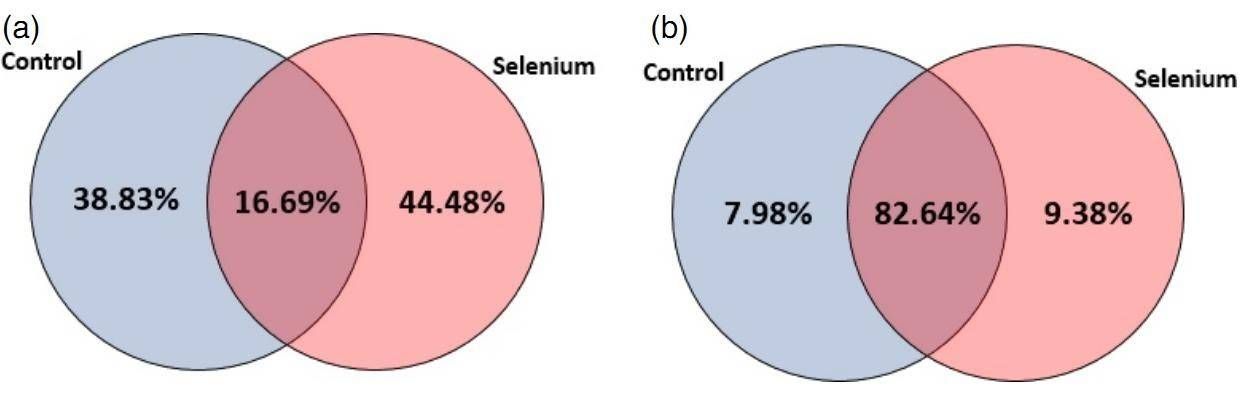 Figure 1. Distribution of miRNAs between control and Se treatment: (a) conserved miRNAs; (b) novel miRNAs.
Figure 1. Distribution of miRNAs between control and Se treatment: (a) conserved miRNAs; (b) novel miRNAs.
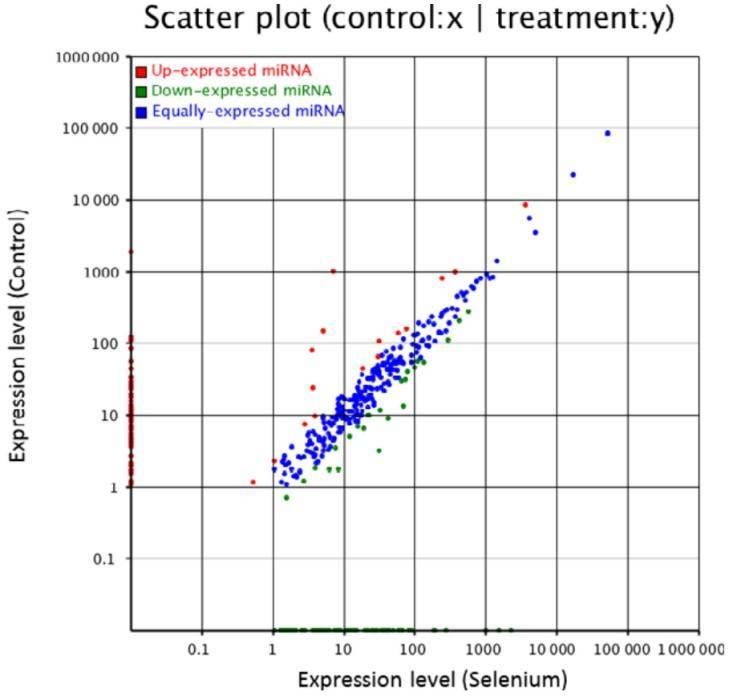 Figure 2. Small RNA expression profiles of control and Se-treated callus of A. chrysochlorus.
Figure 2. Small RNA expression profiles of control and Se-treated callus of A. chrysochlorus.
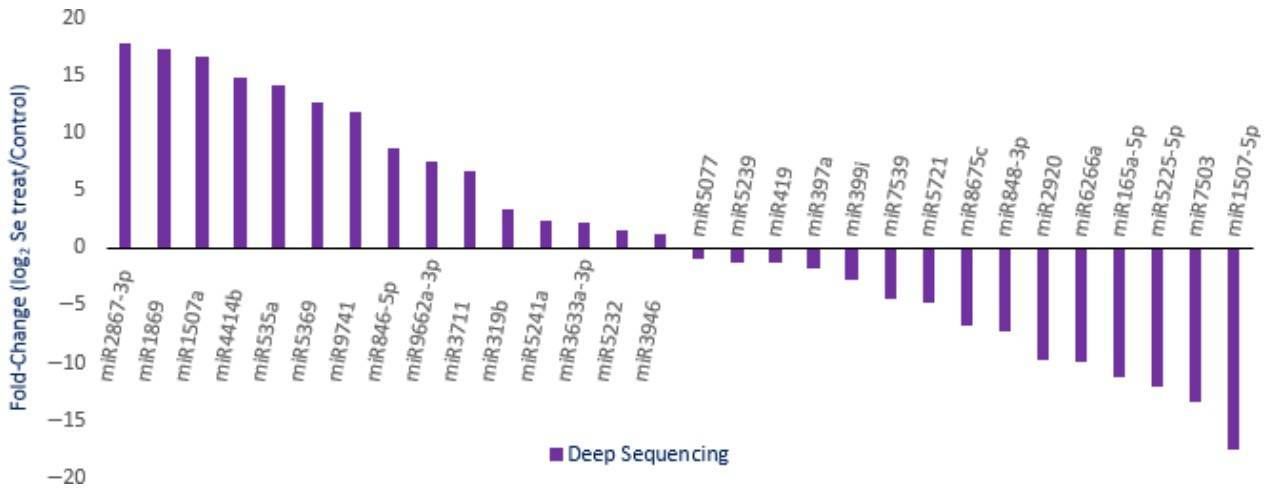 Figure 3. Expression profiles of randomly selected miRNAs with different abundance in Se-treated A. chrysochlorus calli. A total of 1339 predicted sites were identified and determined to be cleaved by 499 miRNAs. The target genes were annotated and classified as transcription factors and their subunits, enzyme coding genes, resistance proteins, leucine-rich repeat, leucine zipper, zinc finger proteins, and other structural and functional proteins.
Figure 3. Expression profiles of randomly selected miRNAs with different abundance in Se-treated A. chrysochlorus calli. A total of 1339 predicted sites were identified and determined to be cleaved by 499 miRNAs. The target genes were annotated and classified as transcription factors and their subunits, enzyme coding genes, resistance proteins, leucine-rich repeat, leucine zipper, zinc finger proteins, and other structural and functional proteins.
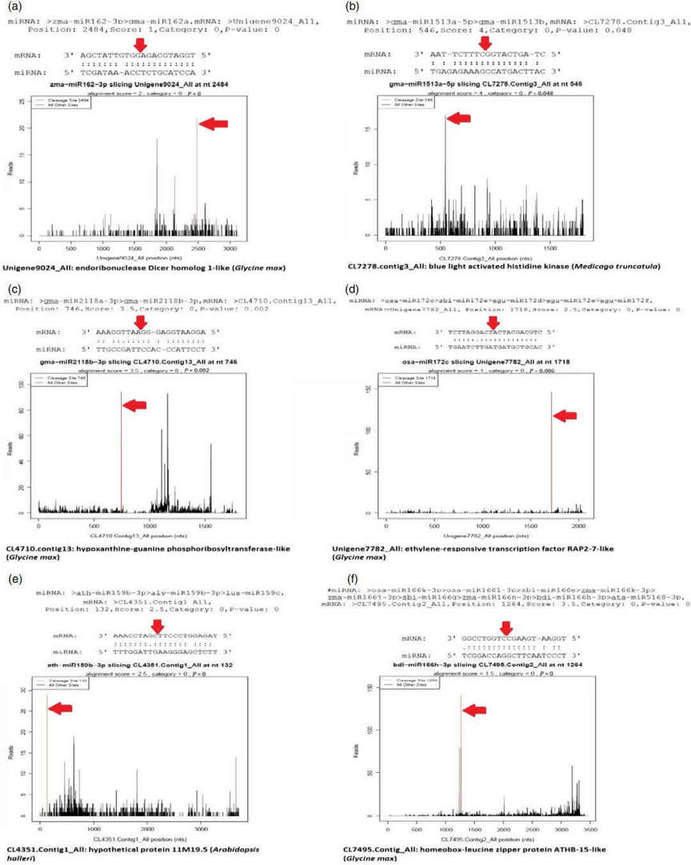 Figure 4. Target plots (t-plots) of miRNAs and their targets. The red arrows indicate the most abundant peaks or cleavage sites. (a) miR162-3p targeting endonuclease Dicer homologue-1-like protein; (b) miR1513a targeting blue light-activated histidine kinase; (c) miR2118b targeting hypoxanthine-guanine phosphoribosyltransferase-like protein; (d); miR172c targeting putative ethylene-responsive transcription factor RAP-2-7-like protein; (e) miR159b-3p targeting hypothetical protein 11M9.5; (f) miR166 h-3p targeting homeobox leucin zipper protein ATHB-15-like protein.
Figure 4. Target plots (t-plots) of miRNAs and their targets. The red arrows indicate the most abundant peaks or cleavage sites. (a) miR162-3p targeting endonuclease Dicer homologue-1-like protein; (b) miR1513a targeting blue light-activated histidine kinase; (c) miR2118b targeting hypoxanthine-guanine phosphoribosyltransferase-like protein; (d); miR172c targeting putative ethylene-responsive transcription factor RAP-2-7-like protein; (e) miR159b-3p targeting hypothetical protein 11M9.5; (f) miR166 h-3p targeting homeobox leucin zipper protein ATHB-15-like protein.
3. GO and KEGG pathway analyses
The targets of identified miRNAs were subjected to GO and KEGG analysis to perceive their biological roles. The target genes are involved in 47 types of cellular component, 103 types of molecular function and 144 types of biological process.
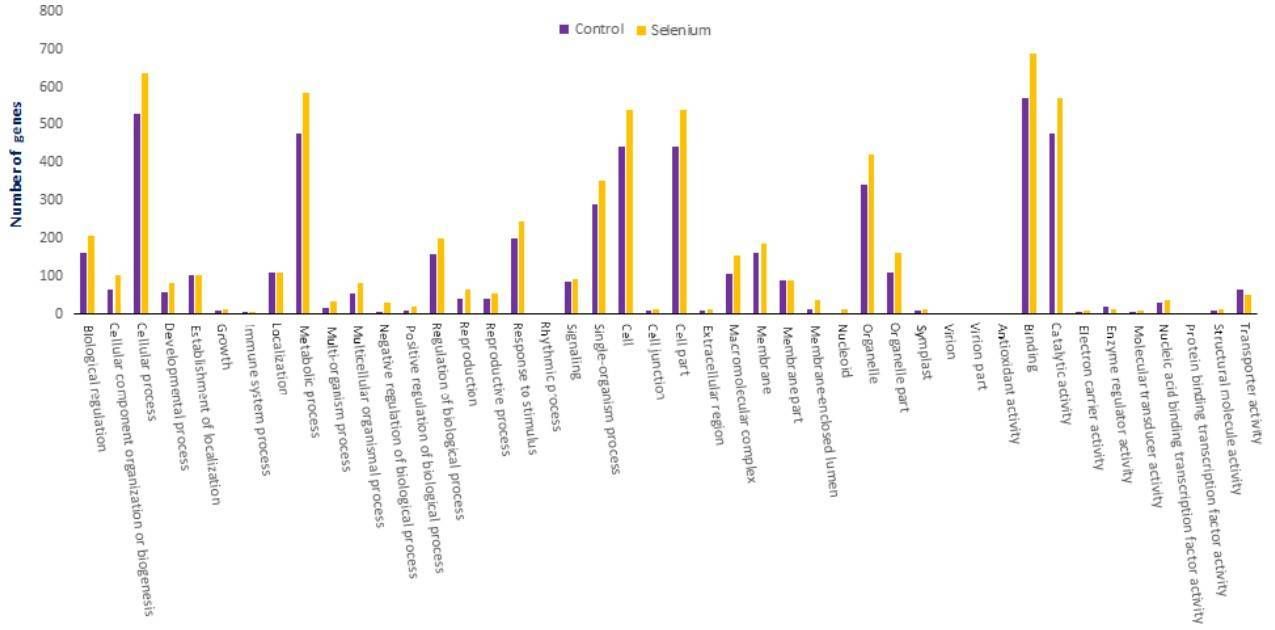 Figure 5. GO classifications of miRNA targets in A. chrysochlorus.
Figure 5. GO classifications of miRNA targets in A. chrysochlorus.
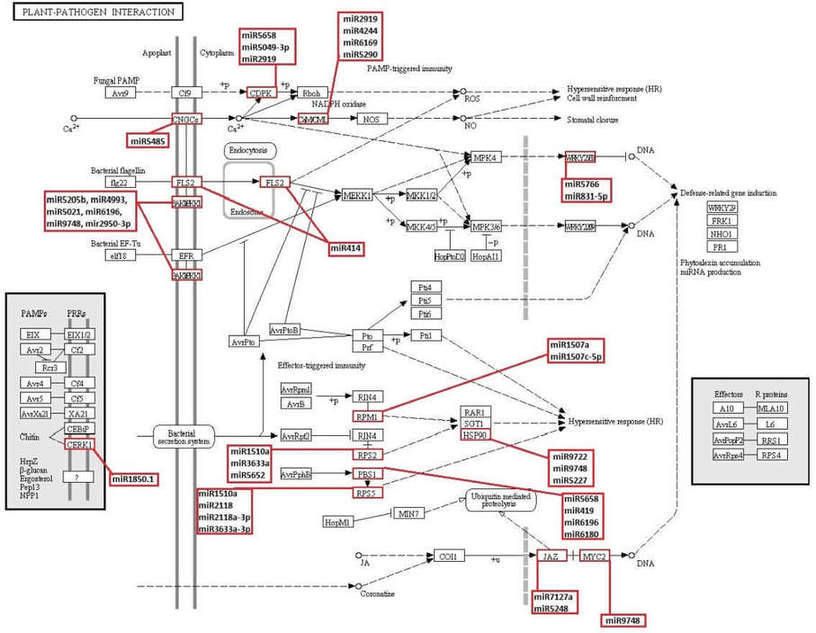 Figure 6. KEGG plant-pathogen interaction pathway and novel and known miRNAs which obtained in this study possibly targeting the genes involved in this pathway.
Figure 6. KEGG plant-pathogen interaction pathway and novel and known miRNAs which obtained in this study possibly targeting the genes involved in this pathway.
Reference
- Cakir O, Candar-Cakir B, Zhang B. Small RNA and degradome sequencing reveals important microRNA function in Astragalus chrysochlorus response to selenium stimuli. Plant biotechnology journal, 2016, 14(2): 543-556.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Chaperone-Mediated Autophagy Controls Proteomic and Transcriptomic Pathways to Maintain Glioma Stem Cell Activity
Journal: Cancer research
Year: 2022
Circular DNA tumor viruses make circular RNAs
Journal: Proceedings of the National Academy of Sciences
Year: 2018
Repeated immunization with ATRA-containing liposomal adjuvant transdifferentiates Th17 cells to a Tr1-like phenotype
Journal: Journal of Autoimmunity
Year: 2024
Role of the histone variant H2A.Z.1 in memory, transcription, and alternative splicing is mediated by lysine modification
Journal: Neuropsychopharmacology
Year: 2024
FAK loss reduces BRAFV600E-induced ERK phosphorylation to promote intestinal stemness and cecal tumor formation
Journal: Elife
Year: 2023
Identification of circular RNAs regulating cardiomyocyte proliferation in neonatal pig hearts
Journal: JCI insight
Year: 2024
See more articles published by our clients.


