
 Sample Submission Guidelines
Sample Submission Guidelines
Whole Genome Re-Sequencing Service
What Is Whole Genome Re-Sequencing
Whole genome re-sequencing is the process of sequencing the genomes of different individuals within a reference genome species, followed by differential analysis of individuals or populations based on the obtained data. Genome resequencing is primarily used to assist researchers in identifying various types of variations such as single nucleotide polymorphisms (SNPs), copy number variations (CNVs), and insertions/deletions (Indels), thus expanding the genetic characteristics of a biological population from a single reference genome at a lower cost. Whole-genome resequencing is widely applied in the study of human diseases and animal and plant breeding, including the detection of pathogenic genes, genetic variations, prenatal screening, personalized medicine, and more.
Whole Genome Re-Sequencing Service Offering
Sequencing. Extraction of genomic DNA, fragmentation, gel recovery of desired DNA fragments (0.2~0.5Kb), adapter ligation, cluster preparation, and ultimately resequencing of the inserted fragments using the Paired-End (Solexa) method.
Data Processing. After sequencing, the obtained reads are processed to filter out adapter contamination and other pollutants. The processed reads are then aligned to a reference genome for quality assessment of sequencing depth, coverage, uniformity, and other output data.
Advantages of Whole-Genome Resequencing
- Provides a high-resolution genomic base sequence map.
- Enables feature analysis of any genome by combining short insertions and longer fragments.
- Identifies disease-causing alleles that may not have been identified through other methods.
- Identifies potential pathogenic variations, providing information for further in-depth research on gene expression and regulatory mechanisms.
Our Service Advantages
- Accelerated Resequencing Analysis
- Customized Analysis Strategies: Based on different sequencing species and protocols, we offer customized options such as selecting reference genome versions, alignment algorithms, and annotation databases.
- Comprehensive Database Integration: We continuously update and integrate multiple databases and versions to provide accurate gene information and annotations.
- Powerful Omics Integration Analysis Capability: By integrating genome resequencing with other technologies such as transcriptome sequencing and methylation sequencing, we expand beyond single-gene variation data.
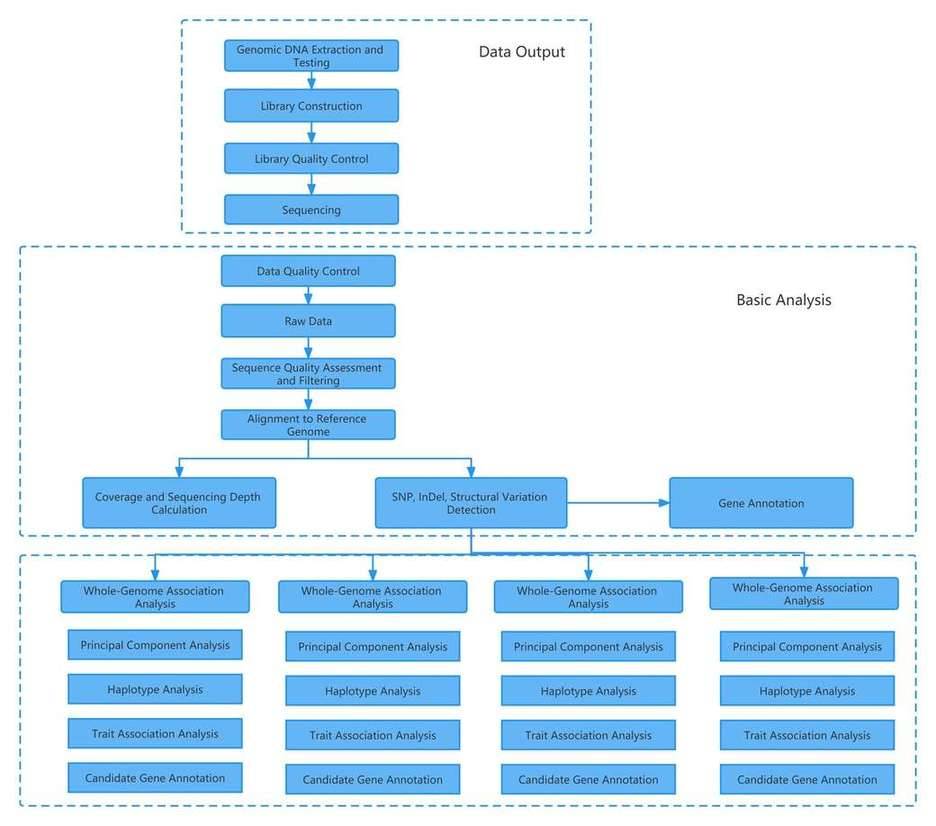
Workflow of Whole-Genome Resequencing
Following standard protocol, the procedures included sample quality assessment, library construction, library quality control, and library sequencing.

Service Specifications
| Sample Requirements Whole Genome Sequencing:
|
|
 Click |
Sequencing Strategies |
 |
Bioinformatics Analysis We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Whole Genome Re-Sequencing for your writing (customization)
Related Solutions
- Cancer Whole-Genome Sequencing: Sequencing of the entire genome of tumors and corresponding normal tissues provides comprehensive insights into the specific mutations associated with cancer. It facilitates the study of oncogenes, tumor suppressor genes, and other risk factors.
- Plant and Animal Sequencing: Whole-genome sequencing of plants and animals is an effective method to explore genes, SNPs, and structural variations while determining the genotype. This information can provide existing gene maps and improve agricultural breeding and screening processes.
- Pathogenic Variation Investigation: Whole-genome sequencing of the human genome enables the identification of disease-related single nucleotide variations and copy number variations. By identifying these pathogenic variations, in-depth genetic research can be conducted targeting pathological features.
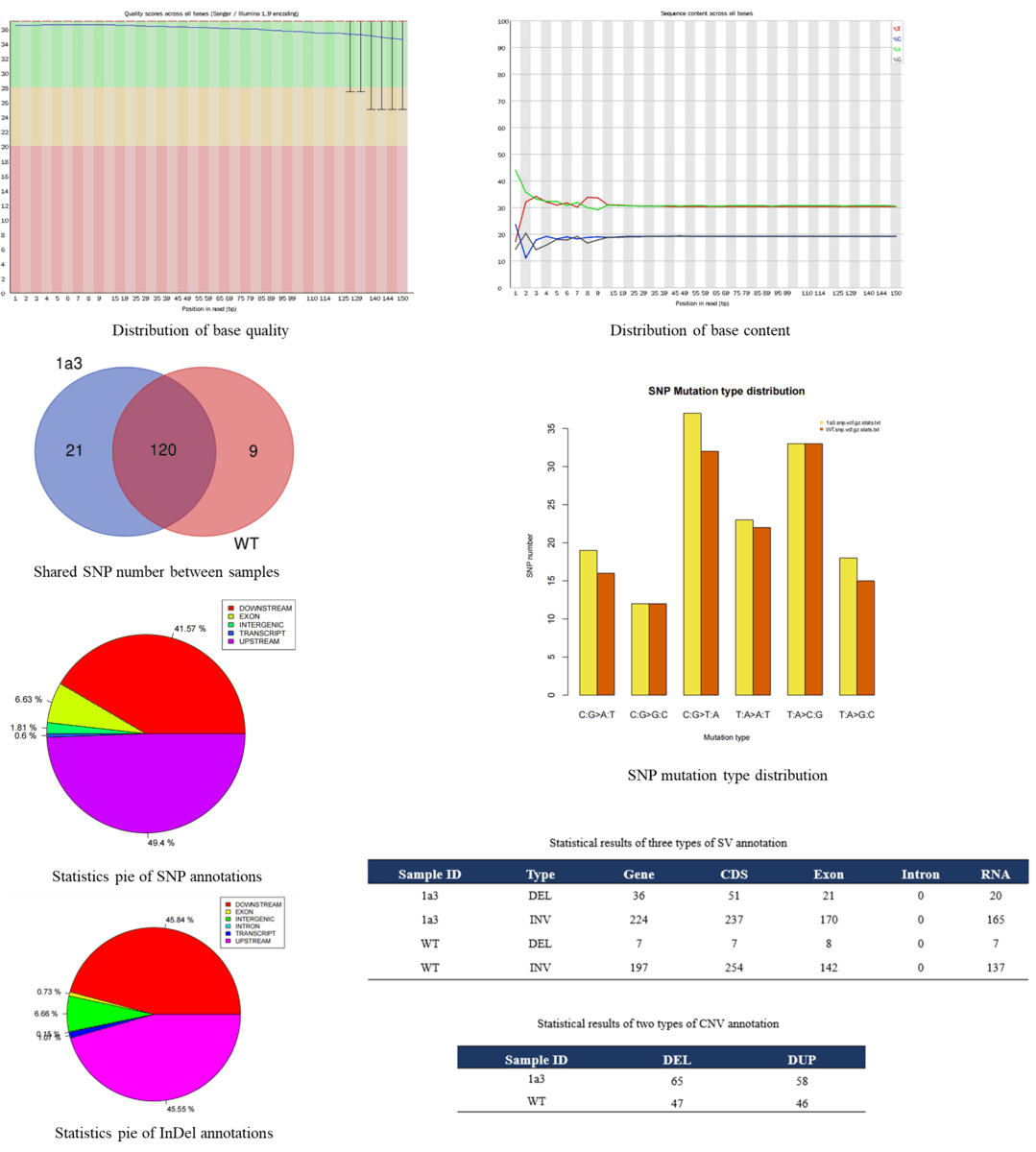
Demo Results
Whole Genome Re-Seq FAQs
1. What are the main applications of whole genome resequencing?
The primary applications of whole-genome resequencing encompass a diverse array of fields in genetic research. These applications include:
- Variant Detection: Identification of genome variations such as Single Nucleotide Polymorphisms (SNPs), insertions/deletions (InDels), and structural variations (SV).
- Disease Research: Unveiling mutations associated with genetic diseases.
- Evolutionary Studies: Analysis of genome disparities and evolutionary relationships among species.
- Population Genetics: Exploration of gene frequencies and genetic structures within populations.
- Crop Breeding: Enhancement of agricultural crop varieties to improve yield and disease resistance.
Furthermore, whole-genome resequencing can be instrumental in tumor molecular subtyping and disease risk screening.
2. What are the differences between second and third generation sequencing for whole genome resequencing?
Whole genome resequencing involves sequencing the entire genome of different individuals within a species to analyze their genetic differences. Second-generation sequencing (e.g., Illumina) reliably identifies SNPs and InDels but faces challenges in detecting and characterizing SVs and in genotyping. Third-generation sequencing, such as Nanopore sequencing, offers read lengths up to megabases (MB), simplifying the resolution of large and complex SVs, and accurately mapping them to the reference genome. It also detects a significantly higher number of variants compared to short-read sequencing.
3. What coverage is required for whole genome resequencing?
Coverage refers to the average number of times each base in the genome is sequenced. Different research objectives require different coverage levels:
- Basic Research: Generally requires coverage of 30× or higher.
- Disease Mutation Detection: Coverage of 50× or higher enhances the accuracy of mutation detection.
- Population Studies: Coverage of 10× to 30× is usually sufficient, depending on the specific research goals.
4. Why is data quality control important in whole genome resequencing?
The meticulous implementation of data quality control (QC) is imperative to safeguard the precision and reliability of sequencing outcomes. Quality control encompasses the following key procedures:
- Eliminating low-quality reads and adapter sequences.
- Assessing data for GC content, base error rates, and depth distribution.
- Ensuring a thorough and consistent genome coverage.
5. What is the cost of whole genome resequencing?
The cost associated with whole genome resequencing is contingent upon variables such as depth of coverage, sample quantity, and the chosen sequencing platform. Typically, the expenditure per sample fluctuates within the range of a few hundred to several thousand dollars. Through progressive developments in sequencing technologies, these costs exhibit a downward trend over time.
Whole Genome Re-Seq Case Studies
Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean
Journal: Genome biology
Impact factor: 13.214
Published: 24 August 2017
Background
Soybean (Glycine max [L.] Merr.) stands out as one of the most significant oilseed and protein crops. The increasing demand for soybean necessitates the enhancement of varieties to boost productivity. However, the interrelationship between different traits and the genetic interactions among genes influencing individual traits pose challenges to soybean breeding. The researchers collected 809 diverse soybean germplasms, cultivated them over two years in three locations, and phenotypically analyzed them based on 84 agronomic traits. Subsequently, whole-genome sequencing (WGS) at a depth of 8.3× was conducted. Through a comprehensive genome-wide association study (GWAS) analysis, the team identified potential genetic loci, interactions among loci, and a genetic network spanning across traits.
Methods
- 809 soybean accessions
- Oil and protein sample preparation
- GC-MS analysis
- DNA preparation
- HiSeq 2500 sequencer
- HiSeq 2000 sequencer
- Read alignment
- Variation calling
- Population genetics analysis
- GWAS
- Construction of association networks
Results
The authors conducted a GWAS on 84 traits using over four million SNP markers from 809 accessions. The analysis controlled for population structure and kinship, resulting in reliable P values and the identification of 150 significantly associated loci (SAL) for 57 traits. Epistasis analysis revealed that the Dt1 locus influences the detection of Dt2 in plant height regulation. By dividing the population based on Dt1 genotypes, additional loci, including Dt2, were identified, confirming previous epistasis findings. The E2 locus was detected in both subgroups, indicating no epistatic interaction with Dt1.
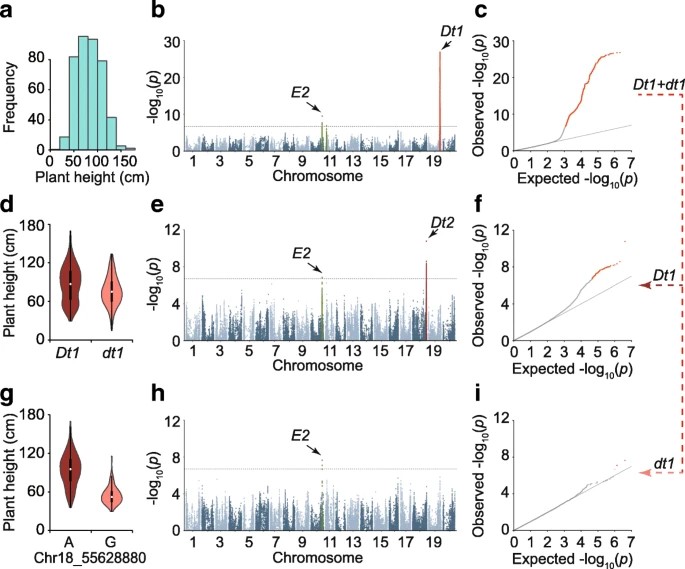 Fig 1. GWAS of the soybean plant height.
Fig 1. GWAS of the soybean plant height.
Soybean is a key oilseed crop, and our study analyzed its genetic architecture related to fatty acid content. The authors identified eight genes associated with fatty acid biosynthesis, including five newly found within SAL regions. Additionally, six genes involved in lipid biosynthesis were linked to fatty acid content. High-fatty-acid alleles correlated with increased total fatty acid (TFA) content, particularly in high-latitude accessions. This suggests an additive function of oil synthesis genes in soybean, similar to maize. Notably, top high-oil cultivars in China lack some high-fatty-acid alleles, indicating potential for developing higher oil content varieties through allele pyramiding.
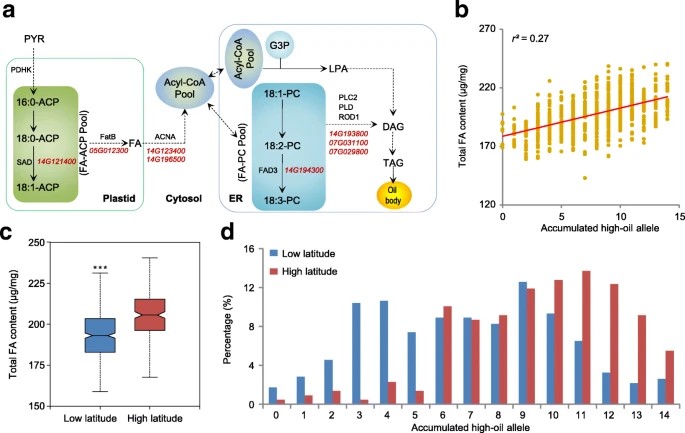 Fig 2. Dissection of genetic regulation of the fatty acid content in soybean.
Fig 2. Dissection of genetic regulation of the fatty acid content in soybean.
The authors observed that the 84 traits related to growth period, architecture, color, seed development, oil content, and protein content were genetically co-regulated, clustering according to their phylogenetic relationships rather than being randomly distributed on the chromosomes. Network analysis indicated that pleiotropy and linkage disequilibrium (LD) contribute to trait correlations, with SAL forming interconnected networks for most traits, except two related to color. Key loci such as E2, E1, Dt1, Dt2, and others were central in regulating multiple traits. For instance, the Dt1 locus influenced not only plant height but also yield-related traits like branch density, stem pod density, stem node number, three-seed per pod, and total seed number.
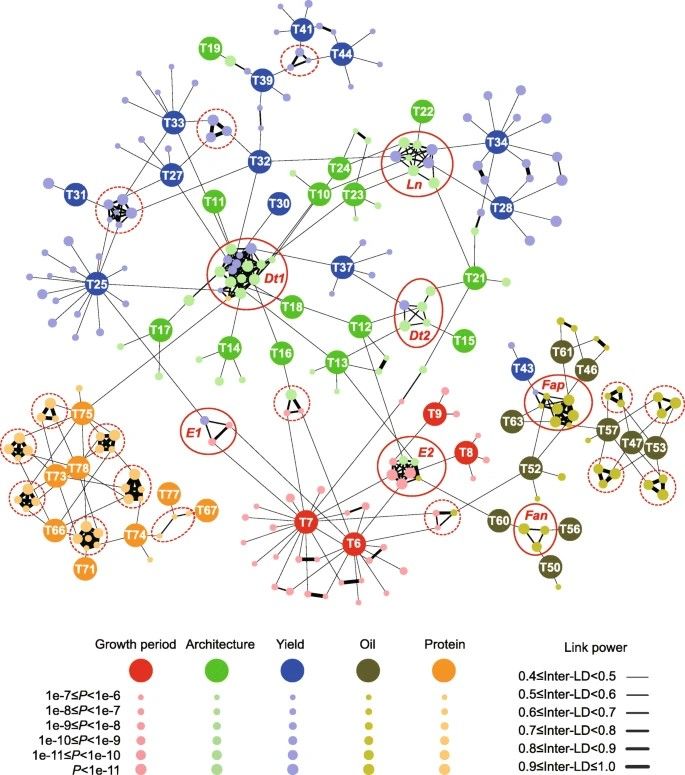 Fig 3. Association networks across different traits in soybean.
Fig 3. Association networks across different traits in soybean.
Conclusion
This study offers valuable insights into the genetic correlations among complex traits, paving the way for future soybean functional research and breeding via molecular design.
Reference:
- Fang C, Ma Y, Wu S, et al. Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome biology, 2017, 18: 1-14.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Distinct functions of wild-type and R273H mutant Δ133p53α differentially regulate glioblastoma aggressiveness and therapy-induced senescence
Journal: Cell Death & Disease
Year: 2024
High-Density Mapping and Candidate Gene Analysis of Pl18 and Pl20 in Sunflower by Whole-Genome Resequencing
Journal: International Journal of Molecular Sciences
Year: 2020
Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI
Journal: New phytologist
Year: 2017
Generation of a highly attenuated strain of Pseudomonas aeruginosa for commercial production of alginate
Journal: Microbial Biotechnology
Year: 2019
Combinations of Bacteriophage Are Efficacious against Multidrug-Resistant Pseudomonas aeruginosa and Enhance Sensitivity to Carbapenem Antibiotics
Journal: Viruses
Year: 2024
Genome Analysis and Replication Studies of the African Green Monkey Simian Foamy Virus Serotype 3 Strain FV2014
Journal: Viruses
Year: 2020
See more articles published by our clients.


