
 Sample Submission Guidelines
Sample Submission Guidelines
What Is Sanger Sequencing?
Sanger sequencing, also known as the “chain termination method,” was developed by the English biochemist Frederick Sanger and his colleagues in 1977. This method is designed for determining the sequence of nucleotide bases in a piece of DNA (commonly less than 1,000 bp in length). Sanger sequencing with 99.99% base accuracy is considered the “gold standard” for validating DNA sequences, including those already sequenced through next-generation sequencing (NGS). Sanger sequencing was used in the Human Genome Project to determine the sequences of relatively small fragments of human DNA (900 bp or less). These fragments were used to assemble larger DNA fragments and, eventually, entire chromosomes.
Sanger Sequencing VS NGS
The development of NGS technologies has accelerated genomics research. NGS can simultaneously sequence more than 100 genes and whole genomes with low-input DNA. Sanger sequencing remains widely used in the sequencing field as it offers several prominent advantages: (i) cost-efficiency for sequencing single genes and (ii) 99.99% accuracy, especially suitable for verification sequencing for site-directed mutagenesis or cloned inserts.
Service you may intersted in
| Feature | Sanger Sequencing | Next-Generation Sequencing (NGS) |
|---|---|---|
| Sequencing Principle | Based on the chain termination method, incorporating special chain-terminating agents to halt DNA synthesis, generating varying lengths of DNA strands. | Utilizes parallel sequencing technologies, simultaneously sequencing numerous DNA fragments, yielding extensive sequence data. |
| Read Length | Relatively long, typically reaching 800-1000 base pairs. | Read lengths vary based on the platform and application but tend to be shorter, e.g., short sequences on the Illumina platform. |
| Speed | Relatively slow, sequencing small DNA segments sequentially, requiring step-by-step analysis. | Relatively fast, capable of concurrently processing multiple samples with high-throughput sequencing. |
| Cost | Relatively high, especially in large-scale sequencing projects. | More economical, particularly in high-throughput sequencing projects. |
| Error Rate | Low, relatively accurate. | High, but errors can be corrected to some extent through repeated sequencing and statistical methods. |
| Applications | Suited for small-scale sequencing projects, SNP identification, gene cloning, etc. | Applicable to large-scale genome sequencing, transcriptome sequencing, protein-nucleic acid interactions, and other high-throughput projects. |
| Equipment Cost | Relatively low, equipment is simpler. | Relatively high, necessitating complex instruments and devices. |
| Scope of Application | Suited for relatively small DNA fragments. | Applicable to large-scale, whole-genome, or whole-transcriptome sequencing. |
| Parallelization | Sequential sequencing, only one DNA strand can be sequenced at a time. | Parallel sequencing, simultaneously processing multiple samples or fragments, enhancing efficiency. |
| Sample Handling | Effective for individual samples, less suitable for high-throughput samples. | Suitable for high-throughput sequencing, capable of processing multiple samples simultaneously. |
| Specificity | Demonstrates good specificity for specific sequences, with minimal cross-contamination. | Some potential for cross-contamination in high-throughput settings, necessitating additional processing steps. |
| Data Analysis | Relatively straightforward, suitable for small-scale projects. | Large-scale data requires sophisticated analysis tools and algorithms. |
| Integration of New Technologies | Mature technology, less adaptable to new technologies. | More adaptable to incorporating new sequencing technologies and methods. |
| Availability | Widely adopted, extensively used in academic research. | Widely applied in academic, medical, and industrial research endeavors. |
How Does Sanger Sequencing Work?
In essence, the underlying principle is as follows:
In Sanger sequencing, a DNA primer complementary to the template DNA (the DNA to be sequenced) is used to be a starting point for DNA synthesis. In the presence of the four deoxynucleotide triphosphates (dNTPs: A, G, C, and T), the polymerase extends the primer by adding the complementary dNTP to the template DNA strand. To determine which nucleotide is incorporated into the chain of nucleotides, four dideoxynucleotide triphosphates (ddNTPs: ddATP, ddGTP, ddCTP, and ddTTP) labeled with a distinct fluorescent dye are used to terminate the synthesis reaction. Compared to dNTPs, ddNTPs has an oxygen atom removed from the ribonucleotide, hence cannot form a link with the next nucleotide. Following synthesis, the reaction products are loaded into four lanes of a single gel depending on the diverse chain-terminating nucleotide and subjected to gel electrophoresis. According to their sizes, the sequence of the DNA is thus determined.
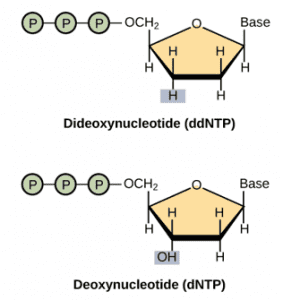
The structure of ddNTP and dNTP.
Image credit: “Whole-genome sequencing” by OpenStax College, Biology).
Illustration through Examples
A canonical DNA amplification system principally comprises four fundamental components: template DNA, primers, deoxyribonucleotide triphosphates (dNTPs), and DNA polymerase. Upon undergoing thermal denaturation treatment, the double-stranded template DNA dissociates into single-stranded oligonucleotides. Employing the principle of complementary sequences, primers bind to these single strands. Leveraging the role of DNA polymerase and the four types of dNTPs as substrates, a complementary DNA strand to the template is synthesized in the 5′ to 3′ direction, thereby yielding a new DNA double helix. By continuously repeating this process, the amplification of the template DNA is achieved.
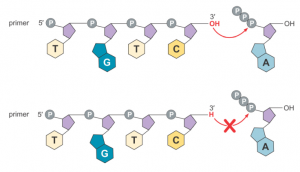
In the DNA amplification system, Sanger introduced a specific factor, denoted as ddNTP. Though ddNTP can be utilized by DNA polymerase as a substrate for DNA strand synthesis similarly to dNTP, it lacks an oxygen atom at the 3′ end. Consequently, it is unable to form a phosphodiester bond with the adjacent deoxyribonucleotide, which ultimately inhibits DNA synthesis at the point of ddNTP incorporation.
Watson et al., MBG, 2008
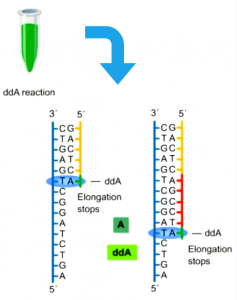
For instance, what transpires when an appropriate concentration of ddT (dideoxythymidine) is introduced into a standard DNA amplification reaction system?
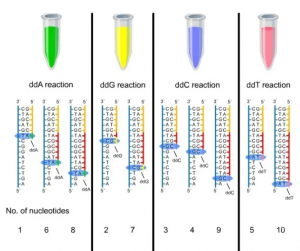
If the template single-strand sequence is 5′-AGTCTAGGCTGAGTC-3′, and the primer sequence is 5′-GACTC-3′, the possible result is the random generation of either a 5′-GACTCAGCCT-3′ sequence or a 5′-GACTCAGCCTAGACT-3′ sequence, when the reaction terminates at the two T points complementary to the template strand A (ddT are italicized).
Please note that it’s imperative to use a suitable concentration of ddT, as an overly high or overly low concentration could impact the preference of newly synthesized DNA strands. For instance, if the concentration is too high, most of the polymerization reactions would terminate at sites closer to the 5′ end, leading to a situation where sequences like (5′-GACTCAGCCT-3′) overwhelmingly dominate. This, in turn, results in errors in subsequent T site detection.
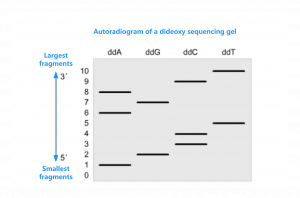
Upon obtaining the newly synthesized DNA sequences, these are usually subjected to gel analysis using high-resolution denaturing polyacrylamide gel. By comparing with the template strand length, one can ascertain the relative positions of the two adenines that complement thymine in the template DNA.
In DNA, there are four types of deoxynucleotides: adenine (A), thymine (T), guanine (G), and cytosine (C). Based on this, Sanger added four different types of dideoxynucleotide triphosphates (ddNTPs) to the DNA amplification reaction system containing the same template strand, sequencing the DNA based on their relative positions within the system. Therefore, the reaction system previously referred to presents itself as follows:
Within the reaction system incorporating ddA/ddG/ddC/ddT, the products are sent for detection using high-resolution denaturing polyacrylamide gel, which consequently enables the assembly of a template strand possessing a complete complementary sequence.
Sanger Sequencing Steps
The Sanger sequencing method consists of 6 steps:
(1) The double-stranded DNA (dsDNA) is denatured into two single-stranded DNA (ssDNA).
(2) A primer that corresponds to one end of the sequence is attached.
(3) Four polymerase solutions with four types of dNTPs but only one type of ddNTP are added.
(4) The DNA synthesis reaction initiates and the chain extends until a termination nucleotide is randomly incorporated.
(5) The resulting DNA fragments are denatured into ssDNA.
(6) The denatured fragments are separated by gel electrophoresis and the sequence is determined.
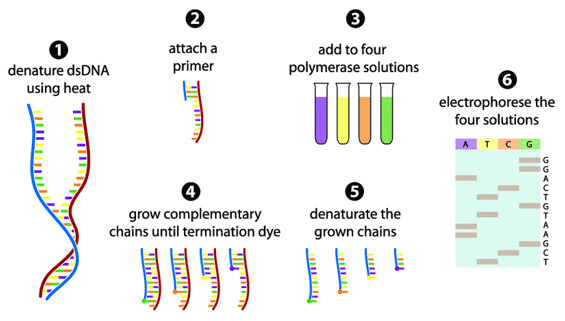
The Sanger sequencing method in 6 steps (adapted from Gauthier 2008).
References:
- Gauthier, Michel G. Simulation of polymer translocation through small channels: A molecular dynamics study and a new Monte Carlo approach. Diss. University of Ottawa (Canada), 2008.
- Sikkema‐Raddatz, Birgit, et al. Targeted next‐generation sequencing can replace Sanger sequencing in clinical diagnostics. Human mutation 34.7 (2013): 1035-1042.
- Sanger F; Coulson AR (May 1975). “A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase”. J. Mol. Biol. 94 (3): 441–8.
- http://smcg.cifn.unam.mx/enp-unam/03-EstructuraDelGenoma/animaciones/secuencia.swf