
 Sample Submission Guidelines
Sample Submission Guidelines
Comprehensive Insights into DNA Methylation Analysis: Sites, Detection Methods, and Current Research Trends
What is DNA methylation
The term DNA methylation refers to the enzymatic process where a methyl group is transferred from S-adenosylmethionine (SAM) to a particular base in the DNA molecule, cytosine (C), adenine (A), and guanine (G) under the action of DNA methyltransferase. This process induces alterations in chromatin structure, DNA conformation, DNA stability, and the interaction between DNA and proteins, thereby modulating gene expression. As a relatively stable form of modification, DNA methylation can be inherited through DNA replication under the action of DNA methyltransferase, making it a critical mechanism in epigenetics. Therefore, DNA methylation provides a unique method of modifying gene activity without altering the DNA sequence itself.
The Principle of DNA Methylation
DNA methylation symbolizes a critical epigenetic modification process. This procedure is catalyzed by DNA methyltransferases, which use S-adenosylmethionine as a methyl donor and selectively append it to the cytosine in the two nucleotides of DNA's CG. The primary outcome of this process is the formation of 5-methylcytosine. Occasionally, this process also results in the production of small quantities of N6-methyladenine and 7-methylguanine.
In the genome, approximately 60% to 90% of CpG sites are subjected to methylation, and the unmethylated CpGs agglomerate into CpG islands. Interestingly, these CpG islands are typically located within the core sequences of structural gene promoters and transcription start sites. This arrangement adds a compelling facet to the landscape of genomic regulation and gene expression.
Notably, DNA methylation can induce chromatin structural changes in corresponding regions of the genome, leading to the disappearance of cleavage sites for nucleases and restriction enzymes within the DNA. This modification results in the chromatin becoming highly spiralled and condensed, thereby losing its transcriptional activity. Furthermore, the 5-position-C-methylated cytosine can transform into thymine under the effect of deamination. This transformation could potentially lead to gene haplotype mutations and base mismatches, like T2G. If such mutations go uncorrected during the cell division process, they could instigate hereditary diseases or cancer.
It is worth emphasizing that methylation in living organisms is a stable and heritable process. This characteristic plays a crucial role in preserving the genetic stability and normal functionality of the cells.
Within the DNA of eukaryotic organisms, 5-methylcytosine is the sole chemically-modified base. Its primary methylation sites are concentrated at the CG dinucleotide clusters, exhibiting a non-uniform distribution throughout the genome, thus designating regions of high methylation, low methylation, and non-methylation. Within mammals, mC constitutes approximately 2-7% of the total cytosine content. DNA methylation represents a significant facet of epigenetic modification, exerting an influence on genetic expression without altering the DNA sequence itself. It forms part of extragenetic coding, and is thus a key mechanism of extragenetic inheritance.
During DNA methylation, a methyl group is added to the DNA molecule, such as at the 5' carbon of the cytosine ring. This 5' methylation pattern is observed in all vertebrates. Roughly 1% of DNA bases within human cells undergo methylation. In mature somatic cells, DNA methylation primarily occurs at the CpG dinucleotide sites, while non-CpG methylation is commonly observed in embryonic stem cells. Within plants, the cytosine methylation can take on the form of symmetrical CpG (or CpNpG) or asymmetrical CpNpNp (where C and G denote bases, p designates a phosphate group, and N stands for any nucleotide).
The methylation status of specific cytosine can be detected via bisulfite sequencing. DNA methylation, if present, can result in gene silencing and consequently lead to dysfunctionality. However, DNA methylation is absent in certain species.
Why is It Critical to Study DNA Methylation
DNA methylation serves a pivotal role in regulating gene expression, hence, investigating it can, at minimum, offer valuable insights into the mechanisms controlling gene expressions. Indeed, DNA methylation is instrumental in numerous vital biological processes, encompassing but not limited to early embryonic development, genomic imprinting, X-chromosome inactivation, silencing of repetitive sequences, alongside the origin, progression, and metastasis of cancer. An in-depth exploration of DNA methylation enables us to comprehend the fundamental mechanisms of these life-sustaining processes more appropriately.
Furthermore, DNA methylation exhibits significant applications within medical research, particularly as a biomarker for cancer. For example, the methylation of Septin9 is broadly employed as a biomarker for colorectal cancer, among others, and its usage is widely acknowledged within clinical practice. This not only exemplifies the potential advantages of DNA methylation in disease diagnosis but also paves the way for its broad future prospects within medical research and potential treatments.
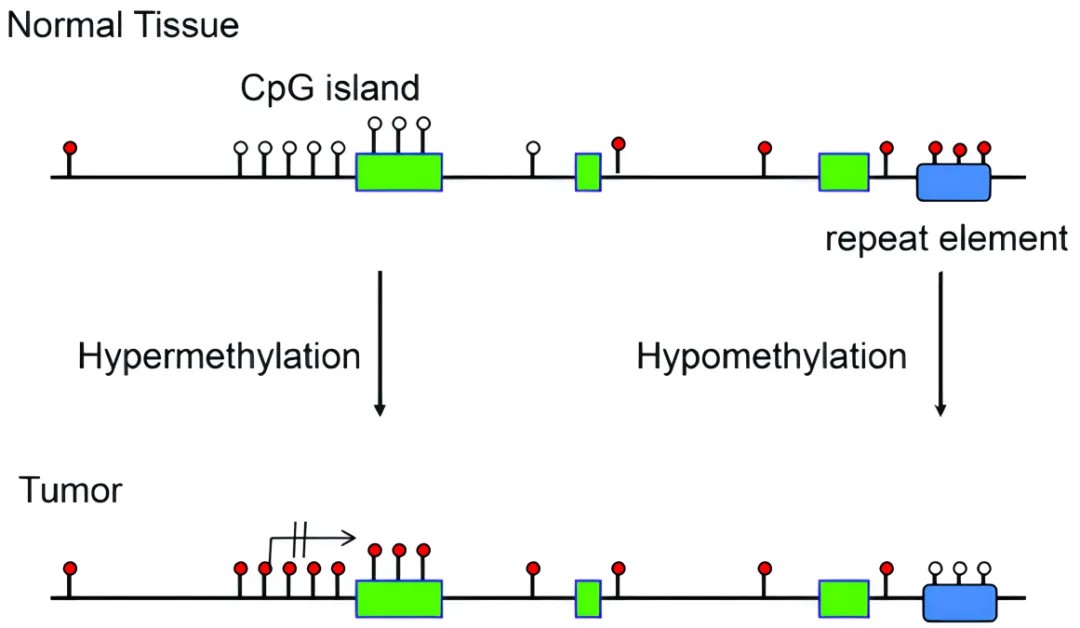 DNA methylation patterns in normal and tumor tissues
DNA methylation patterns in normal and tumor tissues
What is The Extent of DNA Methylation
To delve deeply into understanding human DNA methylation, we need to appraise its fundamental distribution characteristics within the human genome. The genome in humans comprises approximately three billion base pairs, where ~28 million CpG dinucleotides are primary sites of DNA methylation. It is estimated that approximately 60% to 80% of these CpG dinucleotides undergo methylation. Notably, certain areas such as promoter regions contain high-density CpG sites, denominated as CpG islands, which ordinarily do not exhibit methylation. However, in tumor cells, the overall level of methylation markedly decreases, ranging between 20% and 50%. Compared to normal cells, about 10% to 60% of CpG sites in tumor cells exhibit altered methylation states, equivalent to approximately 2.8 million to 16.8 million CpG sites. This alteration suggests that despite a decrease in the overall methylation levels in tumor cells, the methylation levels of CpG sites in the gene promoter regions actually present a clear increase.
How to Analyze DNA Methylation
Brief description of DNA methylation detection methods
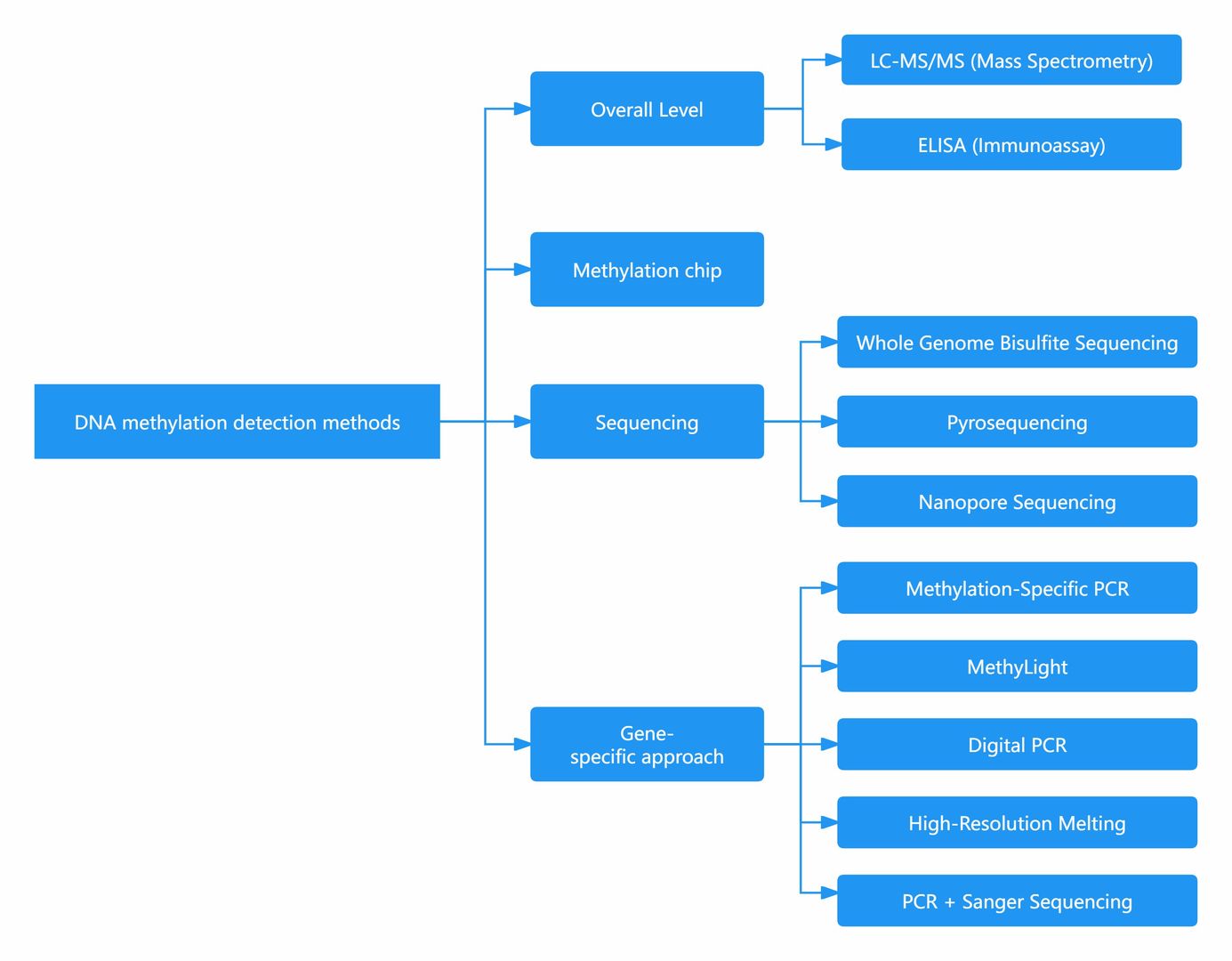
What Technique is Used to Detect DNA Methylation?
| Category | Detection Method | Description | Pros and Cons |
|---|---|---|---|
| Overall Level | LC-MS/MS (Mass Spectrometry) | Accurate, high sensitivity, considered as the gold standard, but high instrument cost and complex operation. | - Accurate - High sensitivity - Gold standard |
| ELISA (Immunoassay) | Fast, low cost, but prone to interference and insufficient sensitivity. | - Fast - Low cost | |
| Methylation chip | Chip-Based Analysis | Suitable only for human samples, ideal for large-scale clinical and targeted DNA methylation analysis, relatively lower cost compared to WGBS. | - Ideal for large-scale clinical and targeted DNA methylation analysis - Lower cost compared to WGBS |
| Sequencing | WGBS (Whole Genome Bisulfite Sequencing) | Comprehensive, able to obtain methylation status of individual bases, preferred for comprehensive understanding of methylation, but complex operation and high cost. | - Comprehensive - Able to obtain methylation status of individual bases - Preferred for comprehensive understanding |
| Pyrosequencing | Able to detect methylation frequency, qualitative and quantitative detection of methylation sites in the sample, suitable for regions within 300bp with few CG sites, but complex operation and short sequencing length. | - Detection of methylation frequency - Qualitative and quantitative detection of methylation sites - Suitable for regions within 300bp with few CG sites | |
| Nanopore Sequencing | Does not require conversion, directly obtains methylation sites and sequences, currently in developmental stage, high cost but likely the future direction. | - No conversion required - Directly obtains methylation sites and sequences - Likely future direction | |
| Gene-specific approach | MSP (Methylation-Specific PCR) | PCR targeting methylated DNA, simple operation, low cost, but primer design can be challenging, suitable for relative quantification of methylation levels in specific gene regions. | - Simple operation - Low cost - Suitable for relative quantification of methylation levels |
| MethyLight | Similar to MSP, uses probe-based PCR, suitable for precise quantification analysis of methylation at specific gene loci or regions. | - Suitable for precise quantification analysis of methylation at specific gene loci or regions | |
| Digital PCR | Similar to MethyLight, but with better sensitivity and amplification efficiency, suitable for low-level methylation detection. | - Better sensitivity and amplification efficiency - Suitable for low-level methylation detection | |
| HRM (High-Resolution Melting) | Similar to MSP, distinguishes methylation based on high-resolution melting curve, fast, but requires high temperature resolution of PCR machine, suitable for detecting highly differentially methylated sites. | - Distinguishes methylation based on high-resolution melting curve - Fast - Suitable for detecting highly differentially methylated sites | |
| PCR + Sanger Sequencing | PCR followed by sequencing analysis, accurately obtains sequence and loci of methylated genes, considered as the gold standard for confirming methylation sites and sequences. | - Accurately obtains sequence and loci of methylated genes - Gold standard for confirming methylation sites and sequences | |
| MassArray | PCR after bisulfite conversion, then flight time mass spectrometry detection, methylated and non-methylated fragments have different molecular weights, high throughput and relatively lower cost, but requires conversion and PCR amplification, and has certain requirements for the number of samples and loci per detection. | - High throughput - Relatively lower cost - Differentiates methylated and non-methylated fragments - Requires conversion and PCR amplification - Certain requirements for the number of samples and loci per detection - Flight time mass spectrometry detection |
DNA Methylation Detection Method in Detail
LC-MS/MS
To comprehensively assess the level of methylation across the entire genome, two methods stand out: liquid chromatography-tandem mass spectrometry (LC-MS/MS) and ELISA.
LC-MS/MS involves enzymatically digesting genomic DNA into individual nucleotides, including A, T, G, C, 5mC, and 5hmC. Subsequently, employing the mass spectrometric multiple reaction monitoring (MRM) mode facilitates the quantification of each nucleotide's abundance, thereby determining the level of DNA methylation (5mC%). Additionally, LC-MS/MS can detect 5hmC. Despite its high accuracy, this method demands expensive instrumentation and entails intricate operational procedures. Given its precision and sensitivity, LC-MS/MS serves as the gold standard for assessing genome-wide 5mC content.
While high-performance liquid chromatography coupled with mass spectrometry ensures high accuracy, it also imposes specific instrumental requirements. Moreover, challenges in ensuring the completion of enzymatic reactions during the enzymatic glycosylation radioisotope labeling process introduce uncertainties and escalate costs. Consequently, this method predominantly finds utility in research contexts.
ELISA
Utilizing an antibody-based approach, the quantification of 5mC content is achieved, coupled with the derivation of DNA methylation levels (5mC%) through standard curve calibration. The enzyme-linked immunosorbent assay (ELISA) method offers simplicity and rapidity, facilitated by commercially available assay kits. However, literature suggests that this method is more suitable for detecting relatively substantial differences in methylation levels.
DNA Methylation Sequencing
With the advancement of high-throughput sequencing technologies, we are now capable of analyzing events such as 5'-methylcytosine and histone modifications at the whole-genome level. These methodologies unveil insights beyond the reach of conventional genomic studies, heralding the era of "DNA methylation sequencing." Moreover, the continual decrease in sequencing costs and iterative advancements in sequencing technologies have vastly expanded the repertoire of available DNA methylation sequencing methods.
Presently, prevalent sequencing methods in epigenetics research include whole-genome bisulfite sequencing (WGBS), precise oxidative bisulfite sequencing (oxBS-seq), optimized versions of reduced representation bisulfite sequencing (RRBS/dRRBS/XRBS), single-cell or low-input whole-genome bisulfite sequencing (scWGBS), amplicon-based (hydroxy)methylation sequencing, and (hydroxy) methylation DNA immunoprecipitation sequencing (hMeDIP-seq). These diverse methodologies offer tailored solutions for various research inquiries pertaining to DNA methylation.
Whole-genome Bisulfite Sequencing
WGBS stands as the gold standard in DNA methylation research, facilitating precise detection of methylation status at every cytosine (C) base across the entire genome. This method furnishes vital technical underpinning for elucidating the spatiotemporal dynamics of genomic DNA methylation modifications, thereby finding extensive utility in unraveling the mechanistic intricacies underlying individual development, aging, disease etiology, and beyond. It emerges as the preferred approach for methylome studies across diverse species.
Conventional whole-genome methylation sequencing techniques entail the utilization of T4-DNA ligase, which, following fragmentation of genomic DNA by ultrasonication, facilitates the ligation of adapter sequences at both ends of DNA fragments. Subsequent treatment with bisulfite converts unmethylated cytosines (C) to uracils (U), which are subsequently converted to thymines (T) via PCR amplification mediated by adapter sequences.
This article on 'How to Choose DNA Methylation Sequencing Technology' will help you select the most appropriate DNA methylation testing method.
Key Advantages:
Broad Applicability: Suitable for research in humans and the majority of animal and plant species (given known reference genomes).
Genome-wide Coverage: Enables comprehensive acquisition of methylome data, facilitating precise mapping of methylation patterns across the entire genome.
Single-base Resolution: Affords the ability to precisely discern the methylation status of each individual C base.
Simplified Methylation Sequencing (RRBS/dRRBS/XRBS)
RRBS is a strategy that employs restriction enzymes to digest the genome, enriching for significant epigenetic regulatory regions such as promoters and CpG islands, followed by bisulfite sequencing. This technique significantly boosts the sequencing depth in high CpG regions, affording high-resolution detection in regions encompassing CpG islands, promoters, and enhancer elements. As a reliable, efficient, and economical tool for DNA methylation research, it holds great prospective applicability in large-scale clinical sample investigations. To adapt to the evolving demands of scientific technology, we have developed dual-enzyme digested RRBS (dRRBS) that can capture CpG loci over broader regions, facilitating the exploration of more expansive methylation areas, including CGI shores.
In an effort to facilitate multi-dimensional analysis with minimal sample volume, we have developed Extended Representation Bisulfite Sequencing (XRBS), a methylation-targeted sequencing method that enriches for coverage of CpG islands, promoters, enhancers, and CTCF binding sites. This method achieves high sensitivity and sample reusability, making it highly scalable and applicable to limited samples and single cells.
Technical Advantages:
High precision: capable of single-base resolution within its coverage range.
Good repeatability: The coverage area's repeatability across multiple samples can reach 85%-95%, making it suitable for differential analysis among multiple samples.
Cost-effective: Sequencing area targets high CpG regions, thereby optimizing data utilization.
Single-cell Whole Genome Bisulfite Sequencing (scWGBS)
The study of DNA methylation genomics in single-cell and minuscule samples is often constrained by library sequencing techniques. Traditional library construction methods and single-cell genome DNA amplification techniques are challenging to apply within methylation experimentation protocols. Using a technology based on linear amplification and single-tube library construction, our team has successfully reduced library bias and precisely completed whole-genome methylation studies in valuable samples.
The research into methylation of single cells and extremely rare samples is primarily applied in exploring the mechanisms of tumor development, cancer research, pre-implantation diagnosis of embryos, early embryonic development, germline cell recombination, stem cells, and cellular heterogeneity, among others. The applicable samples include single cells and micro-cells.
Technical Advantages:
Minimal Starting Materials: Uses single-cell or extremely minimal starting DNA for library construction.
High Sequencing Coverage: Maximizes extraction of comprehensive whole-genome methylation information, accurately mapping the methylation landscape.
Single Base Resolution: Capable of accurately analyzing the methylation status of each cytosine base.
Services you may interested in
- Targeted Bisulfite Sequencing
- Reduced Representation Bisulfite Sequencing
- Whole Genome Bisulfite Sequencing (WGBS)
Learn more
Precise Oxidative Bisulfite Sequencing (oxBS-Seq)
DNA hydroxymethylation has recently been identified as an innovative type of DNA modification that has quickly become a critical research focal point. With edifying research, it has been revealed that bisulfite sequencing, previously touted as the "gold standard" for detecting DNA methylation, is incapable of distinguishing between DNA methylation (5mC) and DNA hydroxymethylation (5hmC). Collaboratively, YeeGene and the University of Cambridge have created an oxBS-Seq that can not only accurately detect DNA methylation by curtailing the influence of DNA hydroxymethylation, but also simultaneously ascertain DNA hydroxymethylation with single-base resolution through utilizing dual-library combination.
Technical Principles: oxBS-Seq oxygenates 5hmC to 5fC, which can then be altered to 'U' by bisulfite, thus enabling precise detection of 5mC. Simultaneously, by comparing with routine bisulfite results, accurate detection of 5hmC can be achieved.
Technical Advantages: Re-defining "gold standard" in DNA Methylation detection Single-base resolution analysis of whole-genome DNA hydroxymethylation modifications Multi-standard validation presents high oxidation efficiency and Bisulfite conversion rate Minimal experimental bias with exceptional reproducibility (R² > 0.98) Capable of accommodating a variety of sequencing application needs: streamlined oxidative Reduced Representation Bisulfite Sequencing (oxRRBS), targeted region oxidative methylation sequencing (Target-oxBS).
Services you may interested in
Learn more
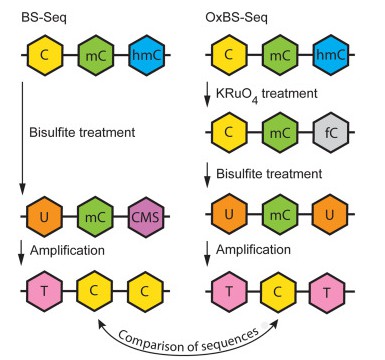 OxBS-seq displays 5mC only and requires a comparison to standard bisulfite sequencing (BS-seq) to infer the sequence positions and abundance of 5hmC and 5mC. (Tibor A. Rauch, et al,. Handbook of Epigenetics, 2023)
OxBS-seq displays 5mC only and requires a comparison to standard bisulfite sequencing (BS-seq) to infer the sequence positions and abundance of 5hmC and 5mC. (Tibor A. Rauch, et al,. Handbook of Epigenetics, 2023)
Amplicon Methylation (Hydroxymethylation ) Sequencing
Amplicon-based methylation and hydroxymethylation sequencing targets specific DNA sequences in the genome by employing methylation-specific amplification primers, such as those relevant to particular genes of interest. Following the (oxidative) bisulfite treatment of genomic DNA, amplification via PCR precedes the profiling of the methylation status within gene regions with an exceptional level of accuracy, facilitated by next-generation high-throughput sequencing.
Technical Advantages:
High Accuracy: This technique is capable of analysing a few to several hundred genes/CpG sites, reaching single-base pair resolution within the targeted range. It further boasts a superior coverage depth (average site coverage >300X).
Cost-Effective: Offering a low cost and rapid turnaround time, this technique brings together outstanding economic efficiency and scientific value.
Specificity: Apt for the detection of methylation states at specific genes or sites.
Broad Applicability: It is widely used in clinical samples for methylation biomarker screening, validation and translational clinical applications.
hMeDIP-Seq
hMeDIP-seq, a term derived from the methylation of DNA, relates to the innovative base modification 5hmC, formed through the oxidation of 5mC by the enzyme produced by the TET family. Not only does 5hmC have the potential to contribute to the control of gene expression by lessening the affinity between the methylated DNA and the Methyl CpG Binding Domain (MBD) of MeCP protein, but it also plays a crucial role in the process of DNA demethylation.
The hMeDIP-Seq technique works by exploiting a 5'-hydroxymethylcytosine specific antibody to enrich hydroxymethylated DNA fragments. This process is then combined with high-throughput sequencing to swiftly and efficiently locate hydroxymethylated regions at the whole-genome scale with a smaller dataset. Its utility is far-reaching, extending to the investigation of the relationship between hydroxymethylation and disease and its role in embryonic development.
This technology presents numerous advantages: a broad detection range that permits the identification of methylated modification areas across the entire genome; a strong specificity that enables targeted detection of regions with methylated modifications within the genome; and a cost-effective advantage with a reduced sequencing dataset hence lowering sequencing costs.
Services you may interested in
Learn more
EM-seq
As for Enzymatic Methyl-seq (EM-seq), it mitigates a major limitation of the long-standing gold standard for methylome analysis, WGBS. The chemical reactions of bisulfite compromise DNA integrity, causing DNA fragmentation and loss. EM-seq overcomes this drawback through enzyme-based processing of DNA for WGBS library construction. In the first step, the TET2 enzyme and oxidation enhancing agents transform 5mC and 5hmC into 5-carboxylcytosine (5caC), thereby safeguarding modified cytosines. In the second stage, the deaminase APOBEC treats cytosines without impacting 5caC, thus allowing the detection of 5mC and 5hmC.
Pyrosequencing
Pyrosequencing, also known as synthetic sequencing, employs bisulfite conversion followed by pyrosequencing to detect DNA alterations within specific target areas after conversion with bisulfite. The quantification of 5mC levels is accomplished by comparing the ratio of cytosine (C) to thymine (T) at individual gene loci. This sequencing technique obviates the need for any special form of fluorescence labeling or electrophoresis, offering ease of operation. With sequencing repeatability and accuracy comparable to Sanger sequencing, pyrosequencing is capable of achieving one hundred times the speed. However, the technique's reliance on the detection of instantaneous luminescence restricts its higher throughput, and its precision in homopolymer detection is suboptimal. As the length of the homopolymer increases, the potential for error correspondingly escalates. Pyrosequencing is most suitable for swift sequencing of short, known DNA sequences ranging from 20 to 50 base pairs.
TET-assisted Pyridineborane Sequencing
TET-assisted pyridineborane sequencing (TAPS) provides an innovative shift from the conventional framework of conversion using bisulfite salts. TAPS opts for a more direct approach, converting methylated cytosines (C) to thymine (T) prior to sequencing. The conversion from C to T is facilitated through a combination of enzymatic and chemical reactions. First, the TET1 oxidation enzyme converts 5mC and 5hmC into 5-carboxylcytosine (5caC). Subsequently, through the action of the reductant pyridine borane, this is transformed into dihydrouracil (DHU), a compound that serves as a PCR template. The DNA polymerase, which can identify uracil (U), recognises DHU. Consequently, after the PCR process, a PCR product that has undergone transformation from C to T is generated.
TAPS offers a distinct advantage: its nondestructive approach towards DNA. It reduces DNA loss, thereby increasing the complexity or coverage of sequencing data. Post-conversion, DNA fragments up to 10 kilobases can be retained, making TAPS highly applicable for third-generation sequencing. Given its method of converting methylated cytosines into thymine, the impact it has on the base balance of the DNA sequence is extremely minimal, which improves the downstream data quality and significantly enhances data alignment rates. Furthermore, the cost-effectiveness of the TAPS technique proves to be superior to the traditional bisulfite conversion methods.
Nanopore Sequencing
Nanopore sequencing technology leverages the potential differences generated when single molecules pass through nanopores for signal detection. The diameter of a nanopore is only sufficient to permit the passage of a singular nucleic acid polymer. The four nucleobases adenine (A), thymine (T), cytosine (C), and guanine (G) - inclusive of those adorned with methylation modifications - each hold distinct charge properties. Consequently, nucleotides bearing methylation modifications and their unmodified counterparts display distinct electrical signal characteristics as they traverse the nanopore. As such, advanced computational models such as deep learning can capture the distinctive electrical signal patterns of methylated molecules, paving the way for the creation of tools capable of detecting DNA/RNA molecular methylation modifications based on ONT sequencing signals.
Services you may interested in
DNA Methylation Chip
In large scale, whole genome DNA methylation studies, the DNA methylation chip method is more viable compared to WGBS due to the high costs associated with sequencing and data processing. A DNA methylation chip involves the hybridization of a bisulfite-treated DNA probe, transforming it into cytosine-enriched CpG loci of interest (both methylated and unmethylated).
The 450K chip performs bisulfite conversion on 0.5-1μg of genomic DNA. The converted DNA then hybridizes with an array of two pre-designed, methylation-specific probes: methylated and unmethylated. The probe's single base binds to labeled and fluorescent-dyed nucleotides (ddNTP) at the 3' CpG site. The bead chip is then scanned on an Illumina iScan to detect the ratio of fluorescent signals. The DNA methylation proportion at each CpG site is termed the β-value (β), computed as M/(M+U+100), where M is the strength of the methylation signal, U is the strength of the unmethylated signal, and 100 is a constant offset used to adjust β-values when the intensities of both methylated and unmethylated cytosines are low. A β-value of 0 signifies 0% methylation, and 1 indicates 100% methylation at the target CpG site.
Methylation chip technology has emerged as a vital tool in DNA methylation research. Compared to whole-genome methylation sequencing, chip detection boasts lower initiation requirements, shorter cycles, and an ability to bypass the limitations of sample type, thus proving applicable to Formalin-Fixed Paraffin-Embedded (FFPE) samples and valuable clinical specimens. Such benefits have made the technology a "hot cake" in the field of methylation detection. Devices like Infinium Human Methylation 450 and Methylation EPIC v1.0 (850K) have been widely harnessed for epigenome-wide association data collection in published studies, showing a steadily increasing trend year after year.
Services you may interested in
Gene-specific approach
Methylation-specific PCR, MSP
MSP, this process allows for a rapid qualitative assessment of the methylation state in any group of CpG sites within CpG islands. The primary advantage of MSP lies in its simplicity and swiftness. If the aim is for relative quantification, one can use fluorescent dye labeling, and through qMSP methodology, directly read fluorescence values and Ct values to represent methylation levels.
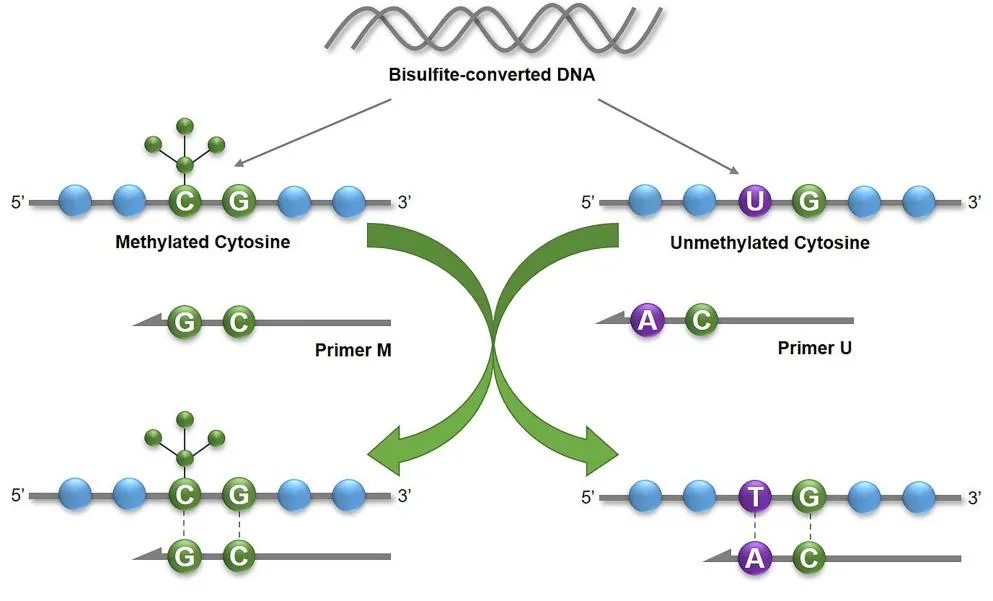 The principle of MSP (J G Herman et al,. 1996)
The principle of MSP (J G Herman et al,. 1996)
MethyLight
Furthermore, methods based on fluorescent probes, such as MethyLight, could be employed. Methylation-specific primers are conjugated with the fluorescent group FAM at the 5' end, while unmethylated-specific primers have VIC, another fluorescent group, conjugated at the 5' end. Real-time PCR releases constant streams of FAM and VIC, allowing the identification of methylation states by detecting these two. The degree of methylation is quantitatively determined by measuring Ct values. Each PCR reaction requires less than 100ng of bisulfite-converted DNA, while the quantitative results can be calculated using standard products.
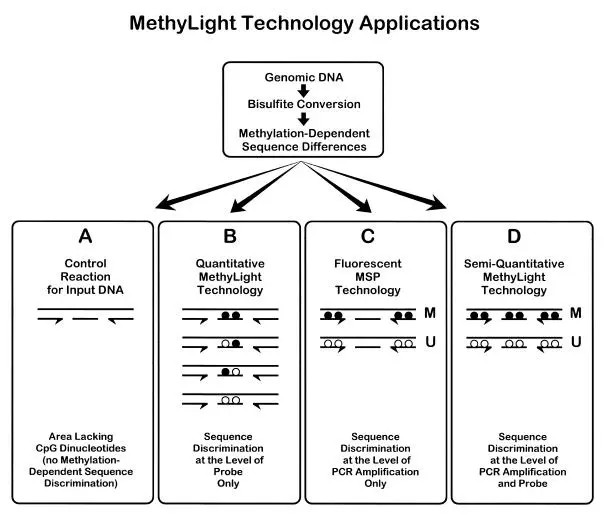 Principle of MethyLight(Anetta Sulewska et al,. 2007)
Principle of MethyLight(Anetta Sulewska et al,. 2007)
dPCR
Digital Polymerase Chain Reaction (dPCR) is a rapidly developing nucleic acid absolute quantification approach based on singularity counting principles. It is progressively employed in quantifying trace or low-level methylation samples. Utilising this method for methylation detection has several distinct advantages, including:
1) High sensitivity: dPCR enables direct methylation detection at a single-molecule level within an extensive array of micro-reaction units, bypassing the need for subcloning vectors required in Sanger sequencing. This is particularly suitable for detecting samples with low abundance or rare methylation sites.
2) Direct quantification: Unlike MethyLight, dPCR negates the necessity of standard curve constructions. It directly quantifies the copy number concentrations of methylated DNA samples, resulting in quantification outcomes less impacted by PCR amplification efficiency.
3) Robust anti-interference ability: By distributing the sample evenly in thousands of micro-reaction units, PCR inhibitors present in the samples are significantly diluted, thereby minimising interference with the PCR reactions.
In light of these characteristics, digital PCR is exceptionally suited for screening and validation of methylation levels in trace DNA samples within complex matrices, such as paraffin-embedded samples or cell-free circulating DNA (cfDNA) samples derived from plasma. Consequently, increasing numbers of researchers are adopting digital PCR techniques in DNA methylation detection.
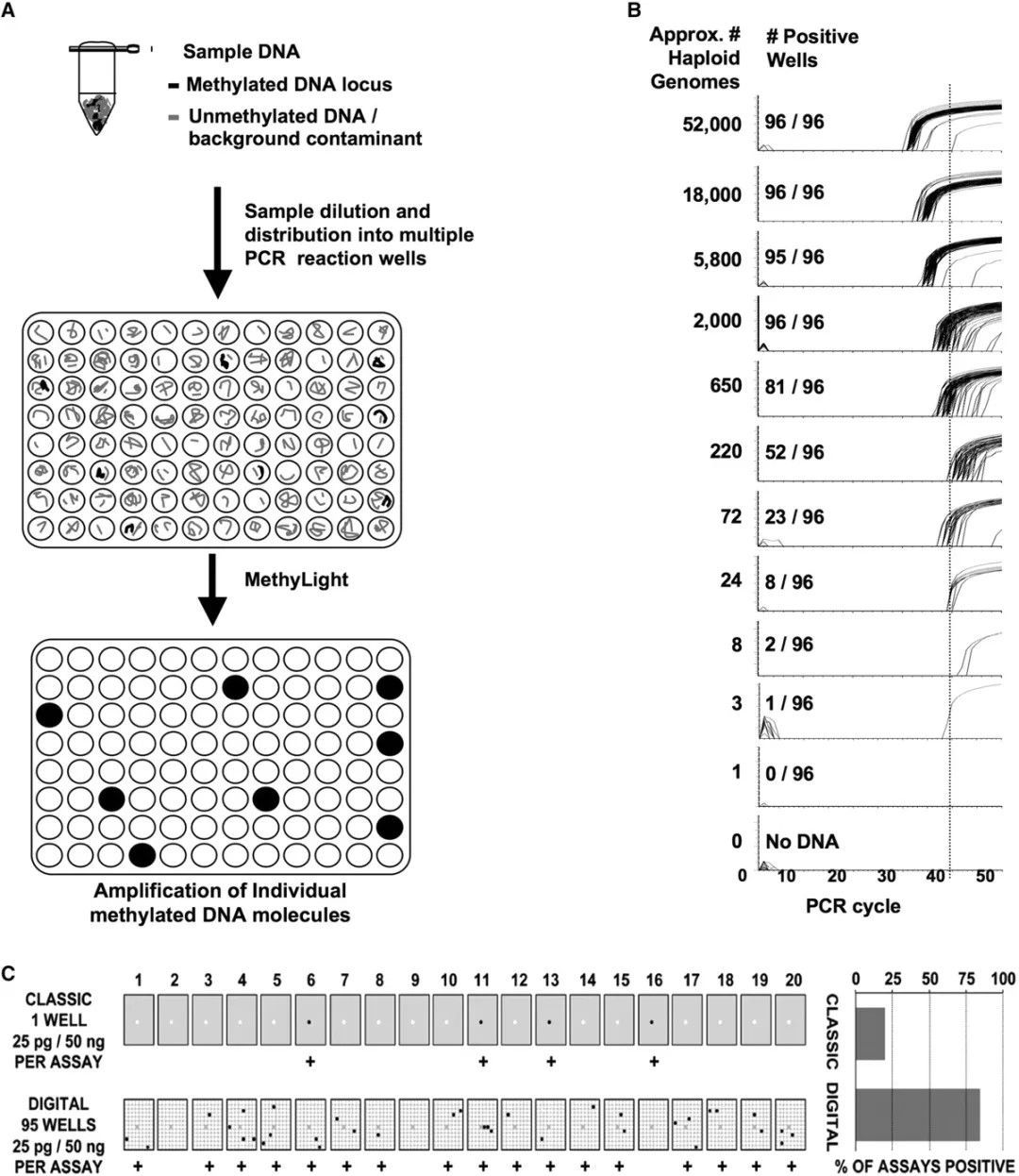 Digital MethyLight-based real-time PCR amplification. (Daniel J Weisenberger et al,. 2008)
Digital MethyLight-based real-time PCR amplification. (Daniel J Weisenberger et al,. 2008)
Methylation Restriction Enzyme Digestion
This method leverages the property of methylation restriction endonucleases to leave methylated regions undigested, thereby fracturing DNA into disparate size segments for subsequent analysis. HpaII-MspI, recognizing the sequence CCGG, is a representative pair of enzymes conventionally employed for this task. Subsequently, isolated products are subjected to Southern blotting or PCR amplification to ascertain the methylation status of the targeted fragments.
Methylated and non-methylated probes, designed respectively with oligonucleotide sections containing hybrid sequences of varying length, are separately hybridized with the test DNA. The probes attached to the DNA are linked by a ligase. If methylation sites exist in the DNA fragment, the probe with the HpaII recognition site cannot be cleaved. On the contrary, if the DNA is non-methylated, the fragment is cleaved by HpaII and the probe cannot be linked. Therefore, in the ensuing PCR amplification process, only the linked probes can be amplified. The final analysis of the amplified fragments reveals the methylation condition of specific sites in the original DNA.
This method boasts several advantages: Firstly, the requirement for sample volume is minimal. Due to the short recognition sequences of the probes, this method is suitable for locoregionally decomposed DNA, such as DNA samples embedded in paraffin. It is also applicable for analyzing large quantities of mixed samples. The limitation of this method lies in the probe linkage site being constrained by the recognition site of the restriction endonuclease. Furthermore, the optimal reaction temperature of the enzyme used needs to be taken into consideration.
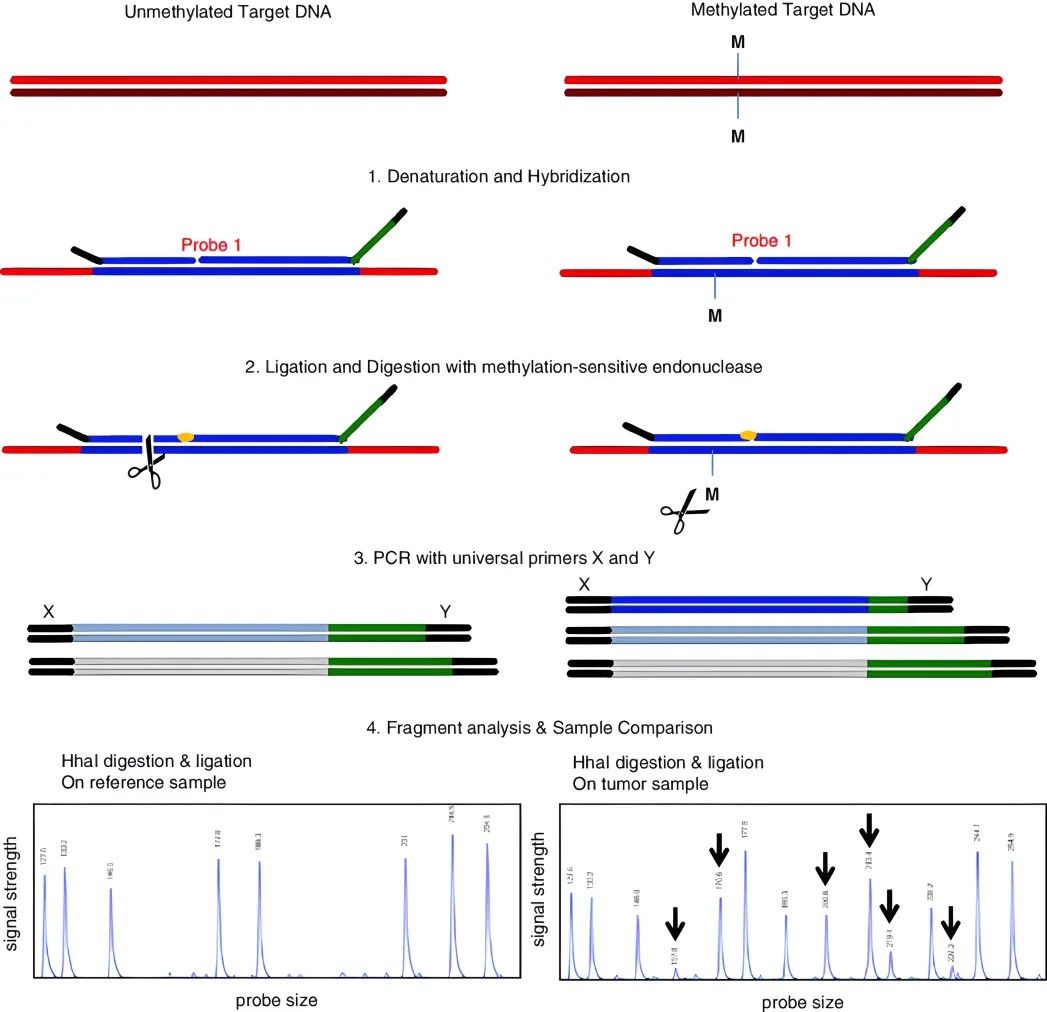 Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA)
Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA)
(Cornelia Hömig-Hölzel et al,. 2012)
HRM
High-Resolution Melting Analysis (HRM) was initially employed for genotyping Single Nucleotide Polymorphisms (SNPs), and it has been extended to detect methylation alterations in DNA subjected to bisulfite treatment. By utilizing specific fluorescent dyes, it identifies the disparities in single bases through their differential denaturation characteristics. The subtle differences between 5-methylcytosine and cytosine present as single-base alterations in DNA following sodium bisulfite treatment. The methylation levels of the test samples can be estimated by comparing their denaturation curves with a series of control curves drawn from samples with known percentages of methylation and non-methylation. This can be achieved with the meticulous design of primers to mitigate PCR bias.
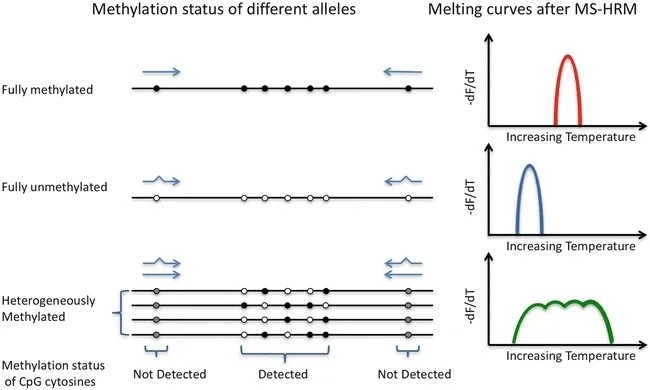 Schematic diagram of high-resolution melting (HRM) detection of methylation (Dianna Hussmann.; Lise Lotte Hansen. 2018)
Schematic diagram of high-resolution melting (HRM) detection of methylation (Dianna Hussmann.; Lise Lotte Hansen. 2018)
Affinity enrichment
Affinity enrichment techniques, including Methylated DNA Immunoprecipitation (MeDIP) and Methyl-binding domain protein capture (MBD Cap), play significant roles in DNA methylation detection. MeDIP employs a 5-methylcytosine-specific antibody to enrich methylated fragments post DNA fragmentation and denaturation. The immunoprecipitated fragments can be sequenced for further analysis post isolation and purification. MBD Cap technique, akin to MeDIP, leverages DNA methylation-binding proteins for the immune precipitation of methylated DNA. Although similar, the enriched proteins differ in that MeDIP generally concentrates on hypomethylated regions with low CpG density, while MBD Cap typically enriches highly methylated regions with high CpG density. These variations make both techniques invaluable in their unique applications in DNA methylation research.
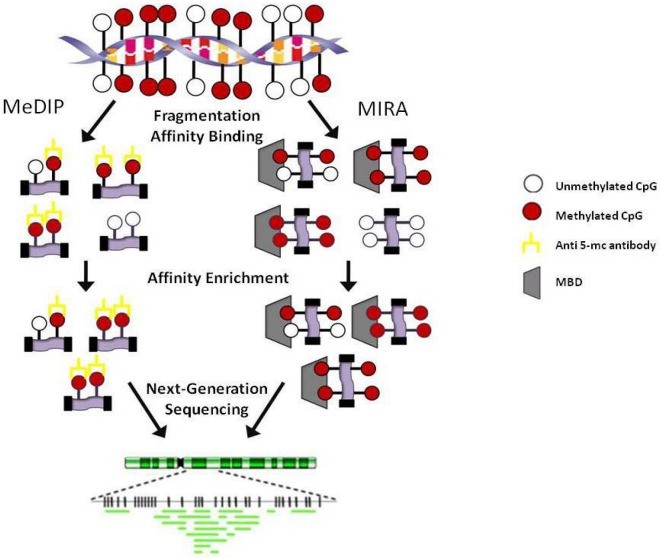 Affinity-based methodologies. MeDIP: Methylated DNA immunoprecipitation; MIRA: Methylated CGI recovery assay; MBD: Methyl-binding domain. (Kristen Taylor et al,. 2010)
Affinity-based methodologies. MeDIP: Methylated DNA immunoprecipitation; MIRA: Methylated CGI recovery assay; MBD: Methyl-binding domain. (Kristen Taylor et al,. 2010)
MassArray
The MassARRAY gene analysis system technology combines the principles of base-specific enzymatic cleavage reaction (MassCLEAVE) and MALDI-TOF sequencing. Similar to other methods detecting DNA methylation, the MassARRAY platform applies sulfite-treated DNA, un-methylated cytosine (C) is converted into uracil (U), whereas methylated cytosine remains unchanged. Methylated and un-methylated cytosines (transformed into U) can be differentiated due to their distinct molecular weights. The resultant distinct molecular weight detection products are transcoded and undergo a base-specific enzymatic cleavage reaction (MassCLEAVE). The methylated and non-methylated fragment molecular weights are different (distinguished based on the 16Da molecular weight difference between base G and base A), enabling methylation ratio calculation through time-of-flight mass spectrometry. MassARRAY offers high throughput, low cost, and high sensitivity. However, special equipment is necessary, and it fails to detect unknown mutations. Moreover, it requires conversion and PCR amplification, and each test has specific requirements regarding sample and site number.
Services you may interested in
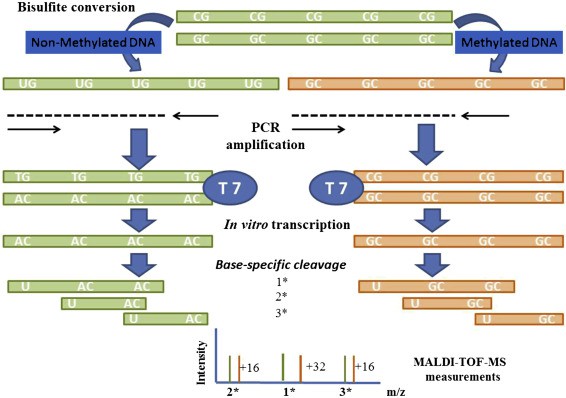 Sequenom MassARRAY Technology for the Analysis of DNA Methylation (Enrique J. Busó et al,. Epigenetic Biomarkers and Diagnostics 2016)
Sequenom MassARRAY Technology for the Analysis of DNA Methylation (Enrique J. Busó et al,. Epigenetic Biomarkers and Diagnostics 2016)
DNA Methylation Research Application
Fundamental Research: Focus on Disease Pathogenesis
a. Cancer: Alterations in the methylation levels of numerous tumor-associated genes lead to gene silencing or overexpression, contributing to tumorigenesis. For instance, in breast cancer, tumor suppressor genes such as BRCA1 and TP53 often undergo DNA methylation, resulting in reduced or lost gene expression, thereby elevating the risk of tumorigenesis. Moreover, aberrant levels of DNA methylation are closely associated with the malignancy and prognosis of tumors. Studies have revealed that highly methylated tumor tissues generally exhibit poorer survival rates and higher metastatic risks compared to tissues with lower methylation levels. Hence, DNA methylation holds significant implications for early screening and diagnosis of cancer.
b. Cardiovascular Diseases: Dysregulated expression of methylated genes and alterations in DNA methylation levels are closely linked to the occurrence of cardiovascular diseases such as hypertension, coronary artery disease, and myocardial infarction.
c. Genetic Disorders: Some monogenic inherited diseases like cystic fibrosis and hemophilia can be diagnosed through methylation.
d. Mental Disorders: DNA methylation may also be implicated in the pathogenesis of mental disorders such as autism and schizophrenia.
e. Autoimmune Diseases: Studies suggest that DNA methylation may play a role in autoimmune diseases like rheumatoid arthritis and systemic lupus erythematosus.
Translational Medical Research: Focus on Disease Biomarkers
a. Cancer Detection: DNA methylation represents a pivotal domain in cancer research, characterized by unique methylation patterns across various cancer types. Assessment of DNA methylation status in cancer tissues can be leveraged for early diagnosis, prognosis evaluation, and recurrence monitoring. For instance, in lung cancer, colorectal cancer, breast cancer, and other malignancies, prediction of patient survival rates and recurrence risks can be achieved through the detection of methylation status in specific genes.
b. Genetic Disease Detection: DNA methylation finds applications in the detection of genetic diseases. For example, examination of the methylation status of certain genes aids in the diagnosis and screening of monogenic inherited diseases like cystic fibrosis and hemophilia.
c. Mental Disorder Detection: Research suggests a potential association between DNA methylation and the onset of mental disorders such as autism and schizophrenia. Profiling the DNA methylation status of individuals with mental disorders contributes to a deeper understanding of their pathogenesis, thereby offering novel insights and approaches for diagnosis and treatment.
In the realm of animal and plant breeding, significant strides have been made, leveraging advancements in molecular biology and environmental science.
In the domain of animal and plant breeding:
Foundational Research: Investigation into the methylation patterns of species and their correlation with various biological phenomena such as growth, development, disease resistance, reproductive capabilities, early embryonic development, and senescence stands as a cornerstone.
Molecular Markers: Utilizing DNA methylation as a molecular marker facilitates breeders in the selection of individuals exhibiting desirable traits such as disease resistance, pest resistance, and high productivity.
Genome Editing: Modulating the methylation patterns of animal and plant genomes enables precise regulation of gene expression, thereby enhancing desired traits in these organisms.
Turning to the environmental aspect:
Phytoremediation: Certain plant species possess the ability to accumulate pollutants. By manipulating the methylation status of these plants, their capacity to absorb and degrade pollutants can be augmented, thereby facilitating phytoremediation efforts.
Microbial Degradation: Harnessing microbial degradation of organic pollutants stands as a pivotal approach in environmental remediation. Manipulating the methylation status of microorganisms can bolster their capability to degrade pollutants.
Molecular Ecological Studies: The exploration of DNA methylation in elucidating the structure, function, and succession patterns of microbial communities in polluted environments aids in a comprehensive understanding of the ecological characteristics of polluted environments, offering a theoretical foundation for pollution management strategies.
References:
- Harrison Aand Parle-Mc DermottA (2011) DNA methylation: a time line of methods and applications. Front.Gene. 2:74.
- DNA Methylation, Methods and Protocols (Second Edition)
- Khodadadi E,Fahmideh L,Khodadadi E,Dao S,Yousefi M,Taghizadeh S,Asgharzadeh M,Yousefi B,Kafil HS.Current Advances in DNA Methylation Analysis Methods. Biomed Res Int. 2021 Mar20; 2021:8827516.
- Ziller MJ, Gu H, Müller F, Donaghey J, Tsai LT, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE, Gnirke A, Meissner A. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013 Aug 22;500(7463):477-81.
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013 Mar;14(3):204-20.
- Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, Cantor CR, Field JK, van den Boom D. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci U S A. 2005 Nov 1;102(44):15785-90.
- Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000 Apr 15;28(8):E32.
- Bendixen, K.K., Mindegaard, M., Epistolio, S. et al. A qPCR technology for direct quantification of methylation in untreated DNA. Nat Commun 14, 5153 (2023).
- Weisenberger DJ, Trinh BN, Campan M, Sharma S, Long TI, Ananthnarayan S, Liang G, Esteva FJ, Hortobagyi GN, McCormick F, Jones PA, Laird PW. DNA methylation analysis by digital bisulfite genomic sequencing and digital MethyLight. Nucleic Acids Res. 2008 Aug;36(14):4689-98.


