
 Sample Submission Guidelines
Sample Submission Guidelines
An Overview of hMeDIP-Seq, Introduction, Key Features, and Applications
For studying 5hmC modifications, hMeDIP-Seq is utilized. It is a small alteration of MeDIP-seq, which is focused on the original MeDIP procedure. Although this technique is theoretically nearly identical to MeDIP-seq, due to the considerably distinct biological knowledge it offers, it is considered as a different procedure. Both alterations of 5mC and 5hmC should be assessed for a detailed understanding into epigenetic changes.
Through immunoprecipitation, methylated DNA is separated from genomic DNA. Anti-5hmC antibodies are subcultured and instigated with fragmented gDNA, preceded by purification of DNA and preparation of a library for sequencing. Deep sequencing offers higher coverage of the genome, showing the majority of hydroxymethylated DNA that is immunoprecipitated.
Advantages and Disadvantages of hMeDIP-seq
The advantages of hMeDIP-seq are as follows:
- In thick and less thick repeat areas, 5hmC is covered.
- Antibody-based selection, due to antibody specificity, is autonomous of sequence and does not enhance 5mC.
On the other hand, its drawbacks include:
- In comparison to single-base resolution with other techniques, the base-pair resolution is lesser (~150 bp).
- In order to prevent non-specific interaction, antibody specificity and selectivity must be evaluated.
- Biased towards areas that are hypermethylated.
Applications and Research of hMeDIP-Seq
DNA methylation at the 5-position of cytosine (5mC) is an essential epigenetic alteration that acts a vital role in the creation of mammals and cell differentiation. Research has shown that the ten-eleven translocation (TET) family of proteins can take away this evidently steady adjustment through oxidation. 5mC-5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) are oxidized by TET proteins (5caC). 5hmC is the most abundant component in vivo among the three 5mC oxidative derivatives and can be identified in almost all mammalian tissues and cells.5hmC is now regarded as an epigenetic alteration, in relation to being one of the medium states of DNA demethylation.
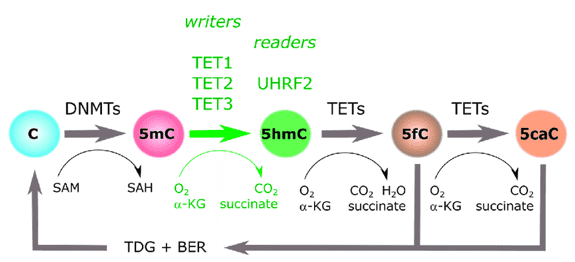
Figure 1. An illustration of the cytosine methylation cycle. (Ecsedi, 2018)
5mC and 5hmC genome-wide mapping uncovers the genomic areas of these alterations, which is crucial for their mechanisms to be elucidated. In order to characterize the genome-wide 5hmC allocation in brain tissues and embryonic stem cells, numerous populations have utilized 5hmC-specific affinity pull-down methods accompanied by next-generation sequencing (NGS) or tiling arrays. Even so, it stays difficult to employ DNA hydroxymethylome information collected by standard hMeDIP-seq to conduct direct genome-wide comparative assessment between high- and low-5hmC specimens because of a few restrictions entombed in the standard hMeDIP-seq procedure. For instance, the existing standard Illumina NGS procedure ensures the loading of the same number of DNA libraries for distinct cluster generation and sequencing specimens, which nullifies the hMeDIP product distinctions between different specimens (especially for those with differential 5hmC abundance).In relation, the various stages of the standard hMeDIP-seq procedure can enhance experimental variance across the specimens, including hMeDIP, library construction, cluster hybridization, and sequencing. Elegant computational techniques have been established by many labs to stabilize or modify the DNA methylome data produced from various specimens. Even so, the prejudice induced by the standard MeDIP-seq or hMeDIP-seq methods' intrinsic restrictions has not yet been overcome. In order to surmount these technical problems, the dramatic variance of 5hmC levels between separate tissues and cells or during cell differentiation and embryo advancement requires innovative strategies and techniques.
The DNA hydroxymethylomes of mouse ES cells (ESCs) and mouse ESC extracted neural progenitor cells were identified and evaluated using hMeDIP-seq in a recent survey (NPCs). The scientists were able to identify different hydroxymethylated areas (DHMRs) between ESCs and NPCs and discovered a complex connection between DNA hydroxymethylation modification and gene expression shifts during ESC neural lineage engagement.
References:
- Ecsedi S, Rodríguez-Aguilera JR, Hernandez-Vargas H. 5-Hydroxymethylcytosine (5hmC), or how to identify your favorite cell. Epigenomes. 2018, 2(1):3.
- Feldmann A, Ivanek R, Murr R, et al. Transcription factor occupancy can mediate active turnover of DNA methylation at regulatory regions. PLoS Genet. 2013, 9(12).
- Tan L, Xiong L, Xu W, et al. Genome-wide comparison of DNA hydroxymethylation in mouse embryonic stem cells and neural progenitor cells by a new comparative hMeDIP-seq method. Nucleic acids research. 2013, 41(7).
- Stroud H, Feng S, Kinney SM, et al. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome biology. 2011,12(6).


