
 Sample Submission Guidelines
Sample Submission Guidelines
Overview of Methylated DNA Immunoprecipitation Sequencing (MeDIP-seq)
What IS MeDIP-Seq
DNA methylation is an epigenetic mark that plays a crucial role in many biological processes, such as embryonic development, gene regulation and disease genesis. The 5-methylcytosine (5mC) is a common methylated form of the DNA base cytosine. The combination of different pretreatment methods followed by different subsequent molecular biology techniques, such as DNA microarrays and next-generation sequencing (NGS), makes DNA methylation mapping feasible throughout the whole genome. Various NGS-based technologies for detecting DNA methylation have been summarized in Table 1 (Su, 2012).
Table 1. NGS based technologies for detecting DNA methylation.
| Pretreatment method | NGS-based analysis | Applications |
| Endonuclease digestion | Methl-seq | Assay a range of genomic elements; Allow a broader survey of regions than classic methylation studies limited to CpG islands and promoters |
| HELP-seq | Measure repetitive sequences, copy-number variability, allele-specific and smaller fragments (<50bp); Detect the hypomethylated loci sensitively | |
| MSCC | Identify unmethylated regions by pinpointing unmethylated CpGs at single base-pair resolution | |
| Affinity enrichment | MeDIP-seq | Generate unbiased, cost-effective and full-genome methylation levels without the limitations of restriction sites or CpG islands; |
| MIRA-seq | Analyze recovered or double-stranded methylated DNA on a genome-wide scale; Be applicable to various clinical and diagnostic situations | |
| MDB-seq | Be applied in any biological settings to identify differentially methylated regions at the genomic scale | |
| MethylCap-seq | Detect differentially methylated regions with high genome coverage; Detect DMRs in clinical samples | |
| Bisulfite conversion | WGBS | Sensitively measure cytosine methylation on a genome-wide scale |
| RRBS | Analyze a limited number of gene promoters and regulatory sequence elements in a large number of samples; Analyze and compare genomic methylation patterns | |
| BSPP | Focus on sequencing the most informative genomic regions; Be applied to Exon capturing and SNP genotyping; Detect methylation in large genomes | |
| BC-seq | Detect site-specific switches in methylation; Determine DNA methylation frequencies in CGIs sampled from a variety of genomic settings including promoters, exons, introns, and intergenic loci |
Methylated DNA immunoprecipitation (MeDIP), which was first described in 2005 (Weber et al., 2005), is a genome-scale purification technique used to enrich methylated DNA fragments with specific antibodies. MeDIP sequencing (MeDIP-Seq) first described in 2008 is a powerful tool used to study 5mC and 5-hydroxymethylcytosine (5hmC) modification by combining MeDIP with high-throughput sequencing (Downet al., 2008). The deep sequencing could provide great genome coverage and reveal the majority of immunoprecipitated methylated DNA.
Services you may interested in
How Dose MeDIP-Seq Work
The workflow of MeDIP-seq is illustrated in Figure 1. It begins with the isolation of methylated DNA fragments by 5mC-specific antibodies via immunoprecipitation. Then the enriched methylated DNA is subjected to DNA purification, library preparation and high-throughput sequencing (Taiwo et al., 2012).
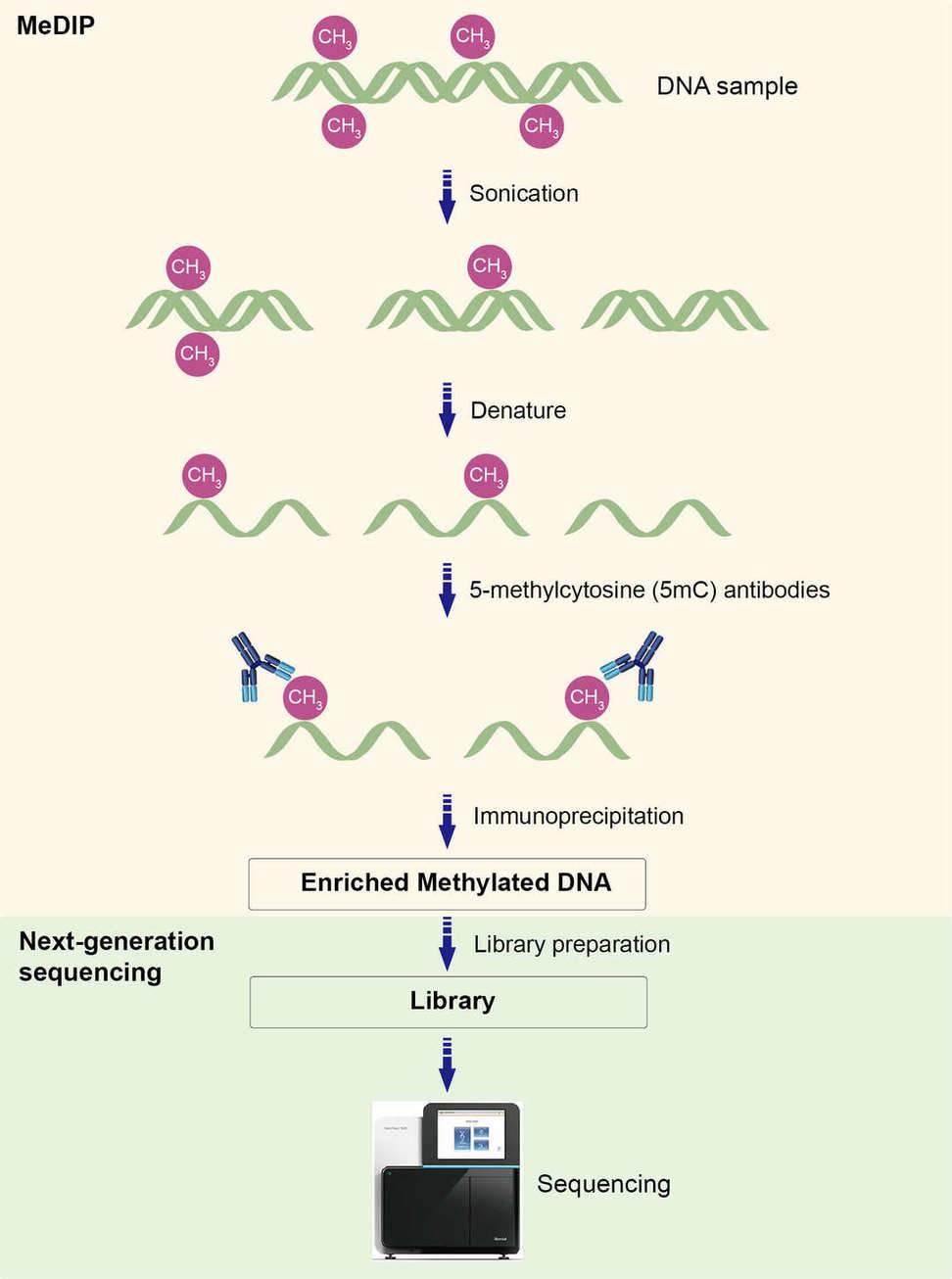 Figure 1. Workflow of MeDIP-seq.
Figure 1. Workflow of MeDIP-seq.
What are the Advantages of MeDIP-seq
- Cover CpG and non-CpG 5mC throughout the genome.
- Be on the Genome-wide scale or in any regions of interest.
- Avoid nonspecific enrichment for 5hmC by using 5mC specific antibody during the process of MeDIP.
- Require low input DNA.
What are the Limitations of MeDIP-seq
- Genomic resolution: resolution of the methylome is lower (~150 bp) compared to single base resolution offered by bisulfite sequencing.
- Nonspecific interaction: antibody specificity and selectivity must be tested to avoid nonspecific interaction.
- Antibody-based selection bias: methylated DNA recovery by the antibody is affected by mC/mCG density, making regions of very low (<1.5%) density may be underrepresented or even interpreted as unmethylated (Taiwo et al., 2012). The antibody-based selection is biased towards hypermethylated regions.
How is MeDIP-Seq Data Analyzed
A commonly used bioinformatics pipeline for analysis of MeDIP-seq data is shown in Figure 2.
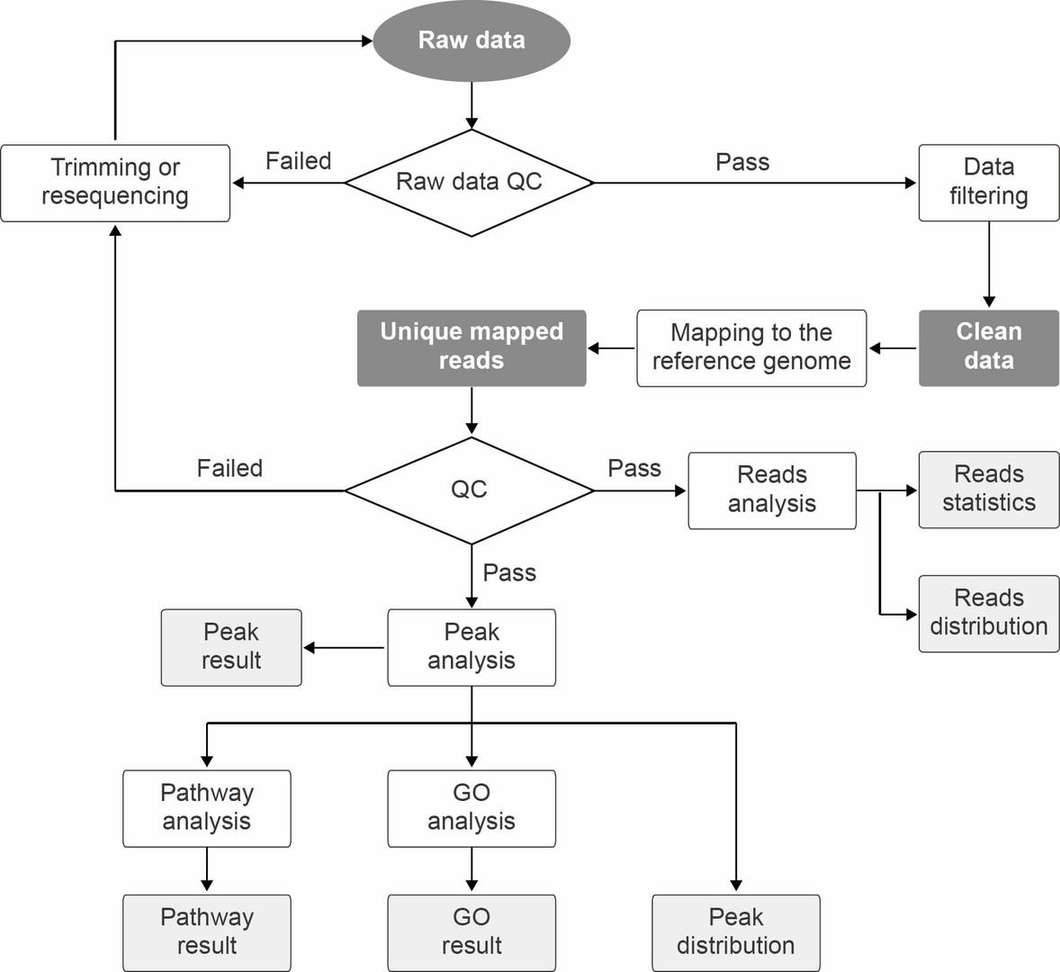 Figure 2. Bioinformatics pipeline for MeDIP-seq data analysis.
Figure 2. Bioinformatics pipeline for MeDIP-seq data analysis.
A number of computational tools have been developed for the analysis of MeDIP data, including Batman, MEDIPS, MeDUSA and MeQA (Table 2). The method to be used largely depends on the purpose of the experiment.
Table 2. Examples of software available for the analysis of MeDIP-seq data
| Software | Summary |
| Batman | Bayesian tool for methylation analysis of MeDIP profiles |
| MEDIPS | Bioconductor package providing a comprehensive approach for normalizing and analyzing MeDIP-seq data |
| MeDUSA | Perform a full analysis of MeDIP-seq data, including sequence alignment, QC and determination and annotation of DMRs |
| MeQA | Pipeline for the pre-processing, quality assessment, read distribution and methylation estimation for MeDIP-seq datasets |
What are the Differences Between Medip-Chip and Medip-Seq
The purified methylated DNA via MeDIP can be input into high-throughput DNA detection methods such as high-resolution DNA microarrays (MeDIP-chip) or high-throughput sequencing (MeDIP-Seq). The workflow overview of MeDIP-chip and MeDIP-Seq is illustrated in Figure 3. In MeDIP-chip, a fraction of the input DNA and the enriched methylated DNA are labeled with cyanine-5 (Cy5; red) and cyanine-3 (Cy3; green) respectively to identify sequences that show significant differences at hybridization levels, thereby confirming the sequence of interest is enriched.
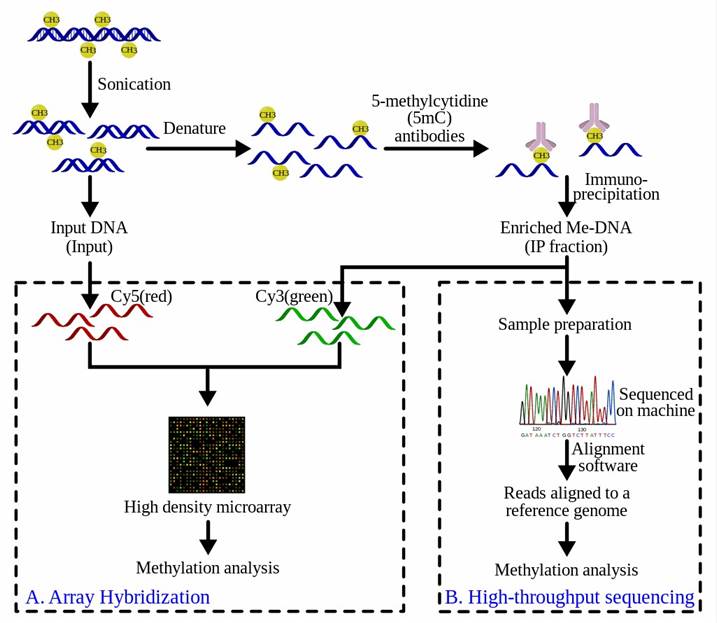 Figure 3. Workflow overview of the MeDIP procedure followed by (A) array-hybridization or (B) high-throughput sequencing (from wiki)
Figure 3. Workflow overview of the MeDIP procedure followed by (A) array-hybridization or (B) high-throughput sequencing (from wiki)
Different DNA methylation analysis approaches have competing strengths and weaknesses. Array-based identification of MeDIP sequences is limited to the array design. As a result, the resolution is restricted to the probes in the array design, whereas the sequence-based identification of MeDIP sequences is generally applicable to any species for which a reference genome exists. A summary of features and potential sources of bias for various techniques is shown in Table 3 (Laird, 2010).
Table 3. Features and sources of bias for various techniques
| MeDIP–chip | MeDIP–seq | RRBS | WGBS | ||
| Features | Unambiguous identification of CpG measured | √ | √ | ||
| In cis co-methylation information | √ | √ | |||
| Non-CpG methylation information | √ | √ | |||
| Allele-specific measurement capability | √ | √ | √ | ||
| Good coverage of regions with low CpG density | √ | ||||
| Compatible with low amounts of input DNA | √ | √ | |||
| Full repeat-masked genome coverage | √ | √ | √ | ||
| Potential sources of bias | Copy-number variation bias | √ | √ | ||
| Fragment size bias | |||||
| Incomplete bisulfite conversion bias | √ | √ | |||
| Bisulfite PCR bias | √ | √ | |||
| Cross-hybridization bias | √ | ||||
| DNA methylation status bias | |||||
| GC content bias | √ | √ | |||
| CpG density bias | √ | √ |
RRBS: Reduced representation bisulfite sequencing
WGBS: whole genome bisulfite sequencing
What are the Applications of MeDIP-Seq
The advantages of cost-effective and requirement for low-input DNA (Taiwo et al., 2012; Zhao et al., 2014) make MeDIP-seq suitable for studies involving low amount of DNA samples, such as oocytes, early embryos (Zhang et al., 2017), and human tumor biopsies (Kim et al., 2011; Zhao et al., 2014).
Analyzing DNA Methylation Patterns: MeDIP-seq can be employed to analyze DNA methylation patterns within the genome, encompassing global patterns and specific gene regions. By observing the methylation spectrum across different samples, changes in methylation can be identified and quantified, unmasking the regulatory mechanisms of DNA methylation in diverse biological contexts.
Identifying Methylation Target Genes: MeDIP-seq can be effectively used to pinpoint methylation genes associated with specific biological processes or diseases. By studying the relationship between methylation levels and gene expression, potential target genes regulated by methylation can be ascertained. This, in turn, enables an in-depth examination of their functionality and control mechanisms in biological phenomena.
Diagnosis and Treatment of Diseases: MeDIP-seq technology plays a crucial role in investigating the contribution of DNA methylation to the genesis and progression of diseases, thereby establishing a foundation for diagnostic procedures, treatment plans, and preventive measures. The comparative analysis of methylation patterns within healthy and diseased tissues can facilitate the identification of associated methylation markers and provide insight into a broad array of diseases. This can help pave the way for groundbreaking therapeutic approaches in the realm of personalized medicine.
Regulation of Methylation in Development and Differentiation: MeDIP-seq can prove instrumental in the exploration of dynamic alterations in DNA methylation throughout development and differentiation processes, thereby shedding light on the function of methylation in determining cellular destiny and influencing tissue development. By closely studying methylation layouts across various stages of development or within specific cell types, it is possible to pinpoint integral methylation events and hence, deepen our understanding of the mechanisms orchestrating development and differentiation.
Interplay of Environmental Factors and Methylation: MeDIP-seq stands as a powerful tool in the inquiry into the repercussions of environmental influences on DNA methylation and in decoding the complex relationship between the environment and epigenetic regulation. Comparisons drawn from the methylation blueprints of samples subjected to varied environmental elements allow clear observation of induced methylation modifications. This provides critical insights for future research into their relevance in environmental adaptation and genetic variability.
At CD Genomics, we are dedicated to providing reliable epigenomics sequencing services, including targeted bisulfite sequencing, reduced representation bisulfite sequencing (RRBS), whole genome bisulfite sequencing, MeDIP sequencing, and ChIP-seq.
References:
- Down, T.A., Rakyan, V.K., Turner, D.J., Flicek, P., Li, H., Kulesha, E., Graf, S., Johnson, N., Herrero, J., Tomazou, E.M., et al. (2008). A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nature biotechnology 26, 779-785.
- Kim, J.H., Dhanasekaran, S.M., Prensner, J.R., Cao, X., Robinson, D., Kalyana-Sundaram, S., Huang, C., Shankar, S., Jing, X., Iyer, M., et al. (2011). Deep sequencing reveals distinct patterns of DNA methylation in prostate cancer. Genome research 21, 1028-1041.
- Laird, P.W. (2010). Principles and challenges of genomewide DNA methylation analysis. Nature reviews Genetics 11, 191-203.
- Su, J. (2012). Advances in Bioinformatics Tools for High-Throughput Sequencing Data of DNA Methylation. Hereditary Genetics 01.
- Taiwo, O., Wilson, G.A., Morris, T., Seisenberger, S., Reik, W., Pearce, D., Beck, S., and Butcher, L.M. (2012). Methylome analysis using MeDIP-seq with low DNA concentrations. Nature protocols 7, 617-636.
- Weber, M., Davies, J.J., Wittig, D., Oakeley, E.J., Haase, M., Lam, W.L., and Schubeler, D. (2005). Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nature genetics 37, 853-862.
- Zhang, Y., Gou, W., Ma, J., Zhang, H., Zhang, Y., and Zhang, H. (2017). Genome methylation and regulatory functions for hypoxic adaptation in Tibetan chicken embryos. PeerJ 5, e3891.
- Zhao, M.T., Whyte, J.J., Hopkins, G.M., Kirk, M.D., and Prather, R.S. (2014). Methylated DNA immunoprecipitation and high-throughput sequencing (MeDIP-seq) using low amounts of genomic DNA. Cellular reprogramming 16, 175-184.


