
 Sample Submission Guidelines
Sample Submission Guidelines
MeDIP-seq vs. RRBS vs. WGBS
DNA methylation, an essential epigenetic process, plays a critical role in gene regulation and various biological processes. CpG density, the frequency of CpG dinucleotides in a given genomic region, has emerged as a crucial factor in DNA methylation analysis. Different molecular techniques have been developed to investigate DNA methylation patterns across genomes. In this article, we aim to compare three widely used methods: Methylated DNA Immunoprecipitation followed by sequencing (MeDIP-Seq), Reduced Representation Bisulfite Sequencing (RRBS), and Whole-Genome Bisulfite Sequencing (WGBS). We analyze their preferences for different CpG density regions and highlight their strengths and limitations in DNA methylation analysis.
CpG Density and DNA Methylation Patterns Across Species
Genomic regions can be classified into different categories based on their CpG density, such as CpG islands (CGIs), CpG shores, CpG shelves, and CpG continental shelves. Previous genome-wide analyses in diverse species, including human, rat, bird, and fish, have shown that the majority of genomic regions belong to the low-density category (1-3 CpG/100bp). High-density regions (>5 CpG/100bp) represent less than 10% of the genome.
Recommended Reading: Identification of DMC, DMR, and DMG in DNA Methylation Analysis.
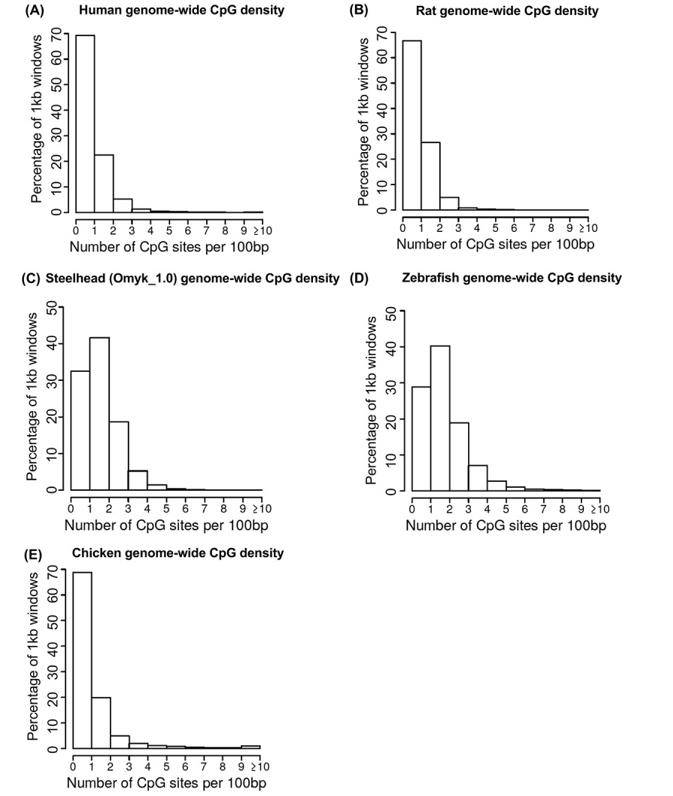 Genome-wide CpG density. (Beck et al., 2022)
Genome-wide CpG density. (Beck et al., 2022)
MeDIP-seq
MeDIP-seq (Methylated DNA Immunoprecipitation followed by sequencing) is a widely used epigenomic technique that enables the genome-wide analysis of DNA methylation patterns at a reduced cost compared to other high-resolution methods. This method involves a series of steps to enrich methylated DNA fragments, followed by sequencing to generate DNA methylation profiles across the genome. MeDIP-Seq uses an anti-5-methylcytosine antibody to enrich methylated DNA regions, followed by next-generation sequencing. MeDIP-Seq predominantly targets low CpG density regions (<5 CpG/100bp), covering more than 95% of the genome. It provides a broad overview of DNA methylation patterns but lacks single-base resolution.
 Methylated DNA immunoprecipitation sequencing (MeDIP-Seq). (Beck et al., 2022)
Methylated DNA immunoprecipitation sequencing (MeDIP-Seq). (Beck et al., 2022)
Despite its utility, MeDIP-seq does have some limitations that researchers should be aware of.
- Lack of Single-Base Resolution
One of the primary limitations of MeDIP-seq is its inability to provide single-base resolution. Since the DNA is fragmented into several hundred base pairs during sonication, the exact location of individual CpG methylation sites cannot be determined. Instead, MeDIP-seq provides information about the overall DNA methylation status within regions containing methylated CpG sites.
- Not a High-Throughput Sequencing Method
Compared to other sequencing methods like Whole-Genome Bisulfite Sequencing (WGBS), MeDIP-seq is not considered a high-throughput technique. This is because it selectively targets methylated DNA fragments, rather than sequencing the entire genome. Consequently, MeDIP-seq may not provide as comprehensive coverage of the genome as WGBS.
- Limited Identification of Individual Levels of CpG Methylation
MeDIP-seq provides information about the presence of methylated DNA fragments but does not directly measure the level of CpG methylation at each site. Consequently, it cannot distinguish between different degrees of methylation (e.g., 50% methylation versus 80% methylation) at individual CpG sites.
- Challenges in Analyzing High-Density CpG Regions
MeDIP-seq may face difficulties when analyzing regions with high densities of CpG sites. The antibody binding efficiency might vary for densely methylated regions, leading to potential biases in the analysis.
RRBS and WGBS
RRBS (Reduced Representation Bisulfite Sequencing) and WGBS (Whole Genome Bisulfite Sequencing) are advanced DNA methylation sequencing techniques known for their ability to achieve single-base resolution.
RRBS is a targeted DNA methylation profiling technique that combines bisulfite treatment with next-generation sequencing. It focuses on CpG-rich regions by using restriction enzymes to fragment genomic DNA before bisulfite conversion, enabling researchers to analyze a representative subset of the genome with higher resolution than MeDIP-seq. RRBS involves a two-step process. Initially, methylation-sensitive restriction endonucleases are used to enzymatically cleave unmethylated DNA at specific CpG sites with high GC density, resulting in DNA fragments. These fragments are then carefully selected based on size and targeted to regions like promoters and CpG islands. Subsequently, the fragments undergo sulfite conversion, which transforms unmethylated cytosine into uracil while preserving methylated cytosine. After this, PCR amplification and sequencing are carried out on these fragments.
 Reduced representation bisulphite (RRBS). (Beck et al., 2022)
Reduced representation bisulphite (RRBS). (Beck et al., 2022)
On the other hand, WGBS involves subjecting the entire genome to sulfite treatment without prior isolation of methylated fragments. WGBS is considered the gold standard for studying DNA methylation as it provides base-pair resolution across the entire genome. It involves bisulfite conversion of genomic DNA, which converts unmethylated cytosines to uracils while leaving methylated cytosines unchanged. Subsequent sequencing allows for precise identification of individual methylated and unmethylated cytosines, providing a comprehensive and detailed picture of the DNA methylation landscape. During sulfite conversion, unmethylated cytosines are converted to uracil, while methylated cytosines remain unchanged. The post-conversion DNA is then sequenced, and diverse bioinformatics protocols are employed, often leading to comprehensive genome characterization.
 Whole genome bisulphite (WGBS). (Beck et al., 2022)
Whole genome bisulphite (WGBS). (Beck et al., 2022)
Comparison of Sequencing Data Results
- MeDIP-seq Analysis
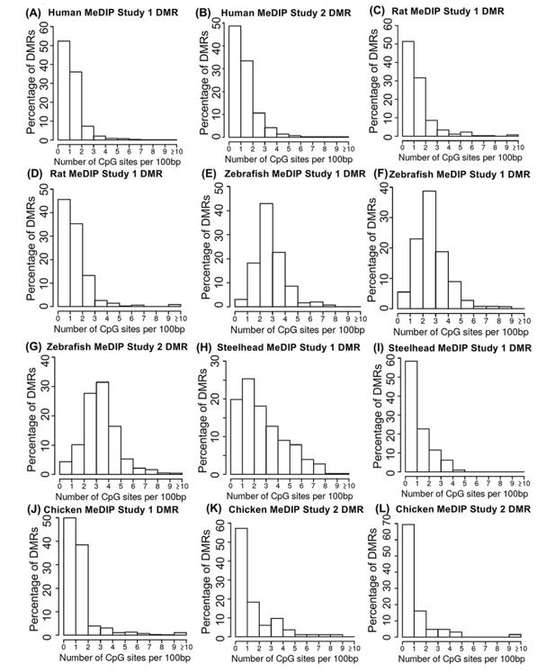
The MeDIP-seq analysis has yielded intriguing findings concerning the distribution of CpG density across diverse species. The data analysis revealed that the majority of differentially methylated regions (DMRs) identified in the MeDIP-seq datasets were primarily associated with regions of low CpG density, specifically within the range of 0-3 CpG sites per 100 base pairs (bp). Notably, a prevalent CpG density of 1 CpG site per 100 bp was observed, which closely correlated with the predominant density within the representative genome.
These outcomes strongly suggest that MeDIP-seq exhibits a proclivity for interrogating genomic regions characterized by low CpG density, thereby effectively capturing changes in methylation occurring in such areas. These regions of low CpG density are widely distributed and encompass over 90% of the genomes of various species. Consequently, MeDIP-seq is a well-suited method for investigating DNA methylation patterns within these regions, thereby providing a comprehensive overview of methylation dynamics across the genome.
Nonetheless, it is noteworthy that some variations in the distribution of CpG density were observed when comparing samples from different species. For instance, the analysis of zebrafish DMRs indicated a shift towards slightly higher CpG densities, specifically within the range of 1-4 CpG sites per 100 bp. This shift may be attributed to the presence of two distinct cell types, spermatozoa and erythrocytes, within the zebrafish samples, each potentially exhibiting unique methylation patterns.
- RRBS Analysis
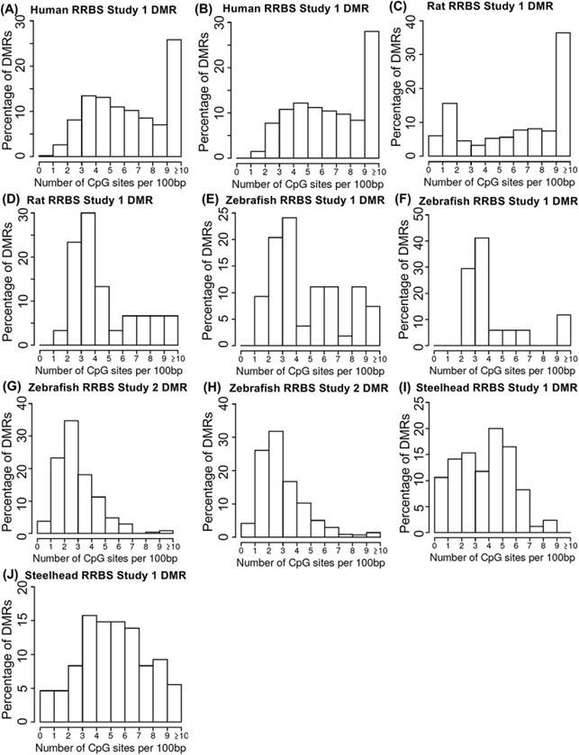
The RRBS (Reduced Representation Bisulfite Sequencing) analysis conducted across diverse species yielded intriguing findings concerning the distribution of CpG density in the acquired datasets. Specifically, differentially methylated regions (DMRs) identified through RRBS exhibited a distinct bifurcation in CpG densities. Certain datasets showcased a shift towards higher CpG densities, particularly in the range of >10 CpG/100bp, while others demonstrated a preference for intermediate CpG densities.
Upon closer examination, it was revealed that within RRBS datasets with CpG densities exceeding 10 CpG/100bp, the dominant density was found to be 10-12 CpG/100bp. Nevertheless, when the analysis was extended to CpG densities over 1 kb, a substantial portion (approximately two-thirds) of the >10 CpG regions fell below 10 CpG/1 kb. This indicates that regions with high CpG density tend to exhibit a decrease in density over longer DNA stretches.
Furthermore, a comparison between MeDIP-Seq and RRBS methods, both applied to the same samples from steelhead trout datasets but conducted by different laboratories, revealed distinct biases associated with each approach. MeDIP-Seq exhibited a preference for lower CpG density regions, while RRBS showed a propensity for higher CpG density regions.
RRBS proves to be particularly well-suited for investigating DNA methylation changes in CpG-rich regions, such as promoters and CpG islands, whereas MeDIP-Seq is more suitable for profiling methylation patterns in low-density CpG regions across the genome. The availability of both MeDIP-Seq and RRBS datasets for the same samples in the steelhead trout study offered a valuable opportunity for cross-validation and highlighted the strengths and limitations of each method.
- WGBS Analysis
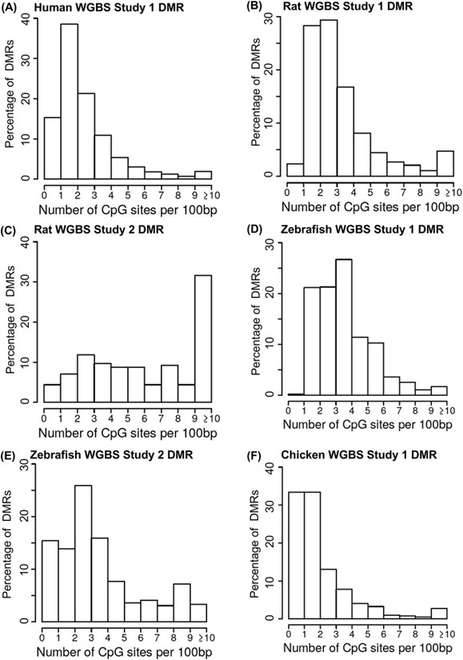
The analysis of whole-genome bisulfite sequencing (WGBS) data across various species has yielded valuable insights into the distribution of CpG density within the acquired datasets. The results reveal that the CpG density in the WGBS dataset falls within a comparable range to that observed in the reduced representation bisulfite sequencing (RRBS) dataset. However, notable differences in the CpG density distributions between these two analyses have been identified.
Specifically, the WGBS datasets exhibit subtle variations, displaying a propensity towards higher CpG densities, particularly in the 2-5 CpG/100bp range and CpG densities exceeding 10 CpG/100bp. This suggests that WGBS data are inclined to target and capture DNA methylation changes in regions enriched with a higher density of CpG sites.
Remarkably, apart from chickens, regions with 1 CpG/100bp were the least detected in the WGBS datasets, implying that WGBS is less likely to identify differentially methylated regions (DMRs) in areas with extremely low CpG densities.
Reference:
-
Beck, Daniel, Millissia Ben Maamar, and Michael K. Skinner. "Genome-wide CpG density and DNA methylation analysis method (MeDIP, RRBS, and WGBS) comparisons." Epigenetics 17.5 (2022): 518-530.


