
 Sample Submission Guidelines
Sample Submission Guidelines
Principles and Workflow of Whole Genome Bisulfite Sequencing
Introduction to WGBS
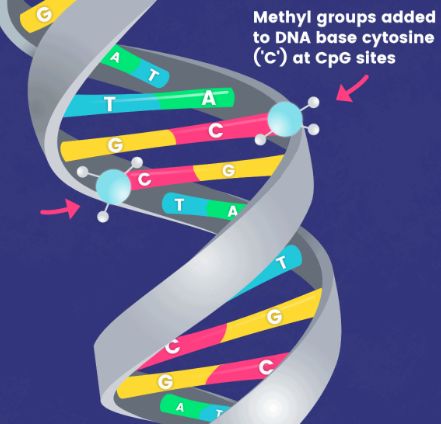
In typical research scenarios, DNA methylation predominantly refers to a methylation process that occurs on the 5th carbon atom of cytosine in CpG dinucleotides, resulting in the formation of 5-methylcytosine (5-mC). This constitutes the main form of DNA methylation in eukaryotes, including plants and animals, and serves as the sole form in mammals. In view of the relative stability of DNA methylation as a modification status, it can be inherited by progeny DNA through the DNA replication process, thus representing a significant mechanism of epigenetic inheritance.
Therefore, the distribution of 5-methylcytosine (or methylome) across the entire genome has garnered considerable attention. Whole Genome Bisulfite Sequencing (WGBS) is a method that utilizes bisulfite treatment to convert unmethylated cytosines (C) in the genome, distinguishing methylated from unmethylated cytosines, coupled with high-throughput sequencing technology to determine methylation status at CpG/CHG/CHH sites. It has been successfully applied in methylome analysis across various branches of eukaryotic phylogeny, multiple species, and in the analysis of methylomes in human embryonic stem cells, induced pluripotent stem cells, peripheral blood mononuclear cells, colon cancer cells, and others. These WGBS datasets have yielded numerous discoveries inaccessible by other methods. With the decreasing cost of sequencing, WGBS is increasingly becoming the method of choice in research. However, traditional WGBS methods pose significant challenges for low-input samples. As the applications of methylation analysis continue to expand, from studies in embryonic development to clinical applications such as early tumor screening, there is a growing demand for methylation library construction from low-input samples.
Services you may interested in
Principles of WGBS
Epigenetic studies have confirmed that DNA-methylation modification of specific gene regions plays an important role in chromosome conformation and gene expression regulation. Methylation of DNA cytosine residues at the C5 (5meC) is a common epigenetic mark in many eukaryotes and is widely found in CpG or CpHpG (H=A, T, C). There are mainly three approaches, including endonuclease digestion, affinity enrichment, and bisulfite conversion (Table 1). Almost all sequence-specific DNA methylation analysis approaches require a methylation-dependent treatment before amplification or hybridization to maintain fidelity. Various molecular biology techniques, such as next-generation sequencing (NGS), are subsequently performed to detect 5meC residues.
Table 1. Main principles of NGS-based methylation analysis.
| Enzyme digestion | Affinity enrichment | Sodium bisulfite | |
| Principles | Some restriction enzymes, such as HpaII and SmaI, are inhibited by 5meC in the CpG. | Affinity enrichment uses antibodies specific for 5meC or methyl-binding proteins with affinity for profiling of DNA methylation. | Sodium bisulfite chemically turns unmethylated cytosine into uracil, hence enabling methylation detection. |
| Method example | Methyl-seq *MCA-seq *HELP-seq *MSCC |
*MeDIP-seq *MIRA-seq |
*RRBS *WGBS *BSPP |
*MCA: methylated CpG island amplification; *HELP: HpaII tiny fragment enrichment by ligation-mediated PCR; *MSCC: methylation-sensitive cut counting; *MeDIP-seq: methylated DNA immunoprecipitation; *MIRA: methylated CpG island recovery assay; *RRBS: reduced representation bisulfite sequencing; *WGBS: whole genome bisulfite sequencing; *BSPP: bisulfite padlock probes.
Various methodologies have been developed to assess the levels of DNA methylation in samples. Bisulfite conversion spurred a revolution in genome methylation analysis in 1990s. Considered the "gold standard" for methylation level determination, WGBS functions on the principle of bisulfite-based methylation analysis. This technique initiates with the treatment of sample DNA with bisulfite, which successfully converts unmethylated cytosine bases to uracil, leaving methylated cytosines unaffected. Subsequent PCR amplification causes uracil to transpose into thymine, distinguishing it from the original methylated cytosines. When coupled with high-throughput sequencing technology, this method enables the mapping of a full-genome DNA methylation profile at single-base resolution.
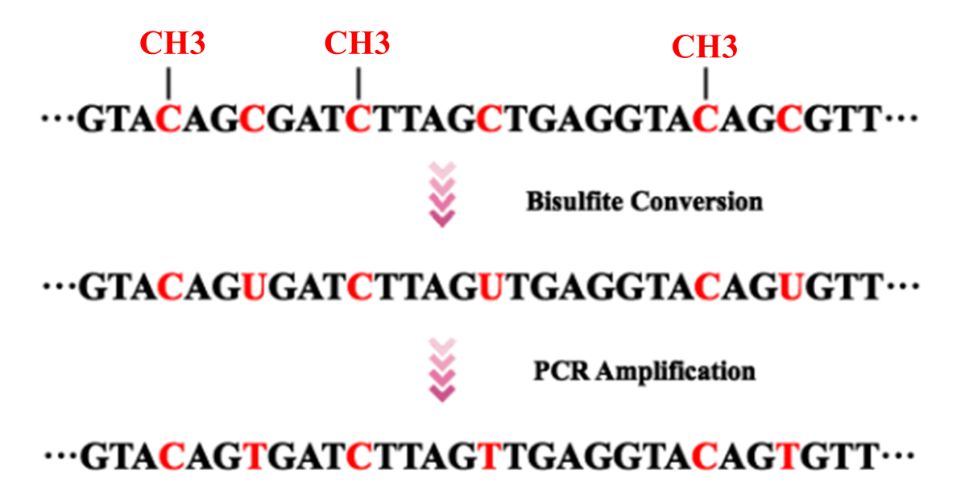 Figure 1. Bisulfite conversion and PCR amplification prior to DNA sequencing.
Figure 1. Bisulfite conversion and PCR amplification prior to DNA sequencing.
WGBS is a high-resolution sequencing technology employed to detect the methylation status of cytosine bases in DNA molecules. Within the framework of WGBS, the DNA sample undergoes bisulfite treatment first, transforming non-methylated cytosines into uracil, whereas the methylated cytosines remain unchanged. Through sequencing analysis, we can determine the methylation status of each cytosine base. WGBS, as a research method of great significance in this field, applies a combination of bisulfite treatment and next/third generation sequencing technologies (mostly, shotgun sequencing) to study DNA methylation at genomic level.
Workflow of WGBS
In short, the basic steps of WGBS include DNA extraction, bisulfite conversion, library preparation, sequencing, and bioinformatics analysis. Here we use Illumina HiSeq as our example to illustrate the workflow of WGBS.
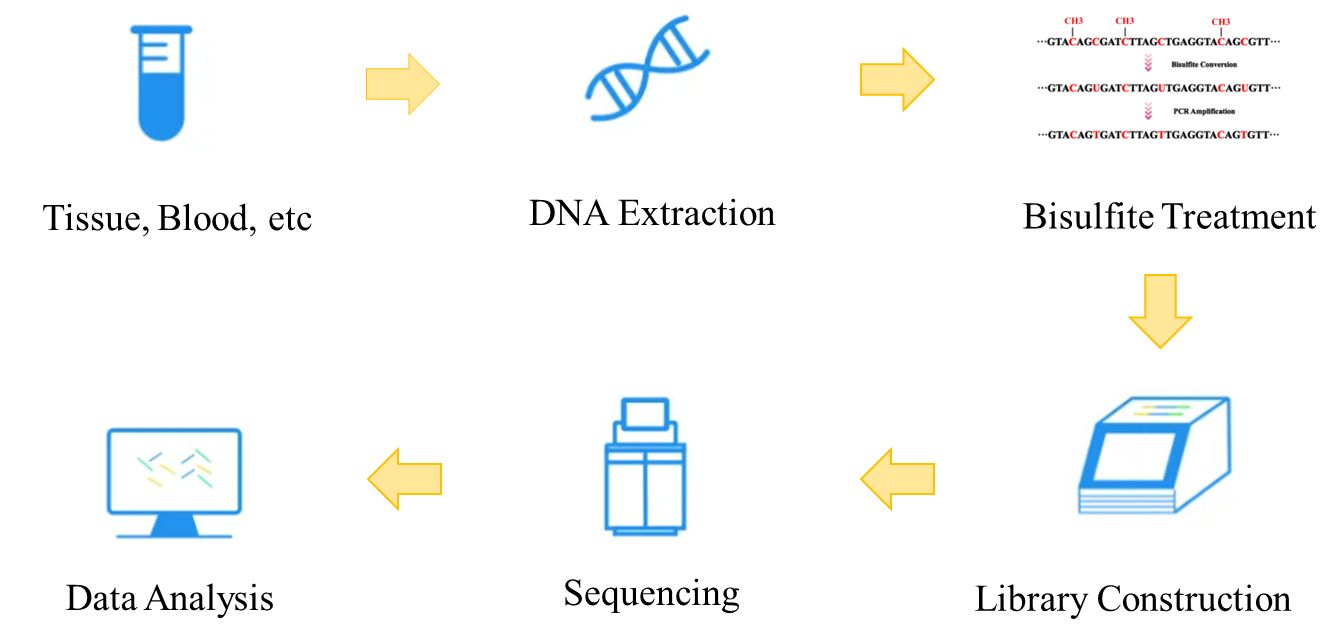 Figure 2. The workflow of whole genome bisulfite sequencing.
Figure 2. The workflow of whole genome bisulfite sequencing.
![Figure 3. The Whole Genome Bisulfite Sequencing Procedure. (Khanna et al. Epignome[trade] methyl-seq kit: a novel post-bisulfite conversion library prep method for methylation analysis. 2013)](https://www.cd-genomics.com/wp-content/themes/v1/images/principles-and-workflow-of-whole-genome-bisulfite-sequencing-3.jpg) Figure 3. The workflow of whole genome bisulfite sequencing (Khanna et al. 2013).
Figure 3. The workflow of whole genome bisulfite sequencing (Khanna et al. 2013).
- DNA Extraction
Firstly, approximately 1-5 mg of tissue samples collected from humans, animals, plants or microorganisms are prepared for DNA. In general, samples for whole-genome bisulfite sequencing need to meet the following four characteristics.
i. Eukaryotes;
ii. Hypomethylation (as shown in Figure 4, studies have shown that once the number of CpG sites in a region increases, the sequencing data of WGBS begins to decrease);
iii. Its reference genome has been assembled to the scaffold level at least;
iv. Relatively complete genome annotations. And then, apply a suitable kit to extract high-purity and high-molecular-weight DNA. The extracted DNA should have a mass of no less than 5 μg, a concentration of no less than 50 ng/μl, and an OD260/280 of 1.8 to 2.0.
 Figure 4. Conventional WGBS technology has low coverage of methylation sites (Raine et al., 2016)
Figure 4. Conventional WGBS technology has low coverage of methylation sites (Raine et al., 2016)
- Bisulfite Conversion
Bisulfite conversion is considered to be the "gold standard" for DNA methylation analysis, the principles have been shown in Figure 5. For this method, BS-induced DNA degradation may lead to depletion of genomic regions enriched for unmethylated cytosines. Therefore, it is important to assess the amount of DNA degradation under reaction conditions, and how this affects the desired amplicon should also be considered. Olova et al. (2018) found that DNA degradation is strong in bisulfite conversion protocols that utilize high denaturation or high bisulfite molarity. There are several kits available in the market (Table 2).
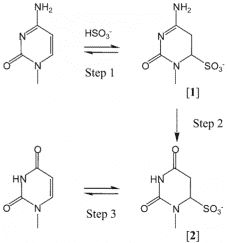 Figure 5. Bisulfite-mediated deamination of cytosine (Hayatsu et al. 2004).
Figure 5. Bisulfite-mediated deamination of cytosine (Hayatsu et al. 2004).
Table 2. Bisulfite conversion protocols and parameters.
| Kits | Denaturation | Conversion temperature | Incubation time |
| Zymo EZ DNA Methylation Lightning Kit | Heat-based; 99 °C Alkaline-based; 37 °C |
65 °C | 90 minutes |
| EpiTect Bisulfite kit (Qiagen) | Heat-based; 99 °C | 55 °C | 10 hours |
| EZ DNA Methylation Kit (Zymo Research) | Alkaline-based; 37 °C | 50 °C | 12-16 hours |
- Library Preparation
Take the EpiGnome™ Methyl-Seq Kit (Epicentre) as an example (as shown in Figure 6), bisulfite-treated single-stranded DNA is random-primed using a polymerase capable of reading uracil nucleotides, to synthesize DNA containing a specific sequence tag. The 3' end of the newly synthesized DNA strand is then selectively labeled with a second specific sequence, thus a two-marker DNA molecular with a known sequence tag at the 5' and 3' ends can be obtained. Illumina P7 and P5 adapters are subsequently added by PCR at the 5 and 3 ends prior to DNA sequencing.
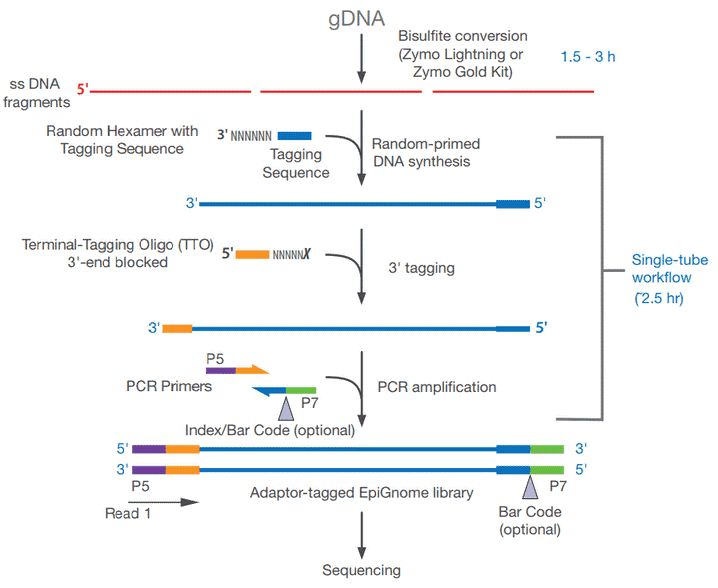 Figure 6. Workflow for the EpiGnome™ Methyl-Seq Kit.
Figure 6. Workflow for the EpiGnome™ Methyl-Seq Kit.
- Sequencing
Hiseq sequencing technology, a novel sequencing method based on sequencing-by-synthesis (SBS), is widely applied for WGBS. The bridge amplification on a flow cell is achieved by using a single molecule array. Since the new reversible blocking technique can synthesize only one base at a time and label the fluorophore, the corresponding laser is used to excite the fluorophore, and the excitation light can be captured to read the base information. Paired-end 150 bp strategy is typically employed in WGBS to sequence 250-300 bp insertion bisulfite-treated DNA libraries. In addition to Illumina HiSeq, PacBio SMRT, Nanopore, Roche 454, and other Illumina platforms are also commonly used for this purpose.
- Data Analysis
A series of analyses can be performed for the sequencing results. Five main types of information analysis are listed in Table 3. In addition, methylation density analysis, differentially methylated region (DMR) analysis, DMR annotation and enrichment analysis (GO/KEGG) and clustering analysis can also be performed. The common bioinformatic resources of WGBS include BDPC, CpGcluster, CpGFinder, Epinexus, MethTools, mPod, QUMA, and TCGA Data Portal.
Table 3. Main types of WGBS data analysis.
| Type | Details |
| Alignment against reference genome | Tools, such as SOAP software, are used to compare the reads with the reference genome sequence, and only the aligned reads will be used for the analysis of methylation information. Align reads allowing C-C matches and C-T mismatches. |
| mC calling | Determine mC position throughout the genome. mC ratios are computed by considering read quality and multi-locus mapping probabilities. Discard small-probability alignment that has a low reliability of alignment. |
| Sequence depth and coverage analysis | An image reflecting the relationship between gene coverage and sequencing depth determines whether methylation discovery can be made with a certain degree of confidence at specific base positions. |
| Methylation level analysis | The methylation level of each methylated C base is calculated as follows: 100*reads/total reads. The genome-wide average methylation level reflects the overall characteristics of the genomic methylation profile. |
| Global trends of methylome | The distribution ratio of CG, CHGG and CHH in methylated C bases reflects the characteristics of whole genome methylation maps of specific species to some extent. |
Advantages and Limitations of WGBS
Advantages :
- Single-base Resolution: The method provides precise analysis down to the single-base resolution, allowing for accurate determination of the methylation status of each cytosine base. It is considered the "gold standard" in methylation level research.
- High Conversion Rate in Methylation Library Construction: The average conversion rate through bisulfite sequencing is above 99%, with strict quality control measures in place during library construction.
- Wide Scope of Application: The method is applicable to methylation research across all species with known reference genomes.
- Full Genome Coverage: It maximizes the acquisition of comprehensive, full-genome methylation information, enabling accurate mapping of the methylation landscape.
- Epigenomic Research: It holds significant potential for studying the spatiotemporal specificity in the field of epigenetics.
Limitations:
- Large data volume required (recommended at least 30x coverage).
- Dependency on a reference genome.
- Elevated AT base content post-treatment, which can impact sequencing and analysis.
- Susceptibility to DNA damage during bisulfite treatment.
Applications of WGBS
(1) Epigenetic Studies: WGBS serves as an instrumental tool for investigating DNA methylation variations among different cell types, tissues, or stages of development, thereby unraveling the role of epigenetic alterations within biological processes. This enhances our understanding of the mechanisms involved in gene expression regulation, cell differentiation, development, and disease occurrence.
(2) Disease Research: In the exploration of disease, WGBS plays a pivotal role. Researchers can contrast DNA methylation patterns in both healthy and disease-affected states, aiming to identify methylation alterations related to disease initiation and progression. Such investigations hold vital significance for studies concerning cancer, neurological disorders, cardiovascular diseases, among others.
(3) Individual Differences and Population Genetics: WGBS also facilitates research into inter-individual DNA methylation variations, aiding in comprehending the genetic variation of methylation within populations. This advances the dissection of the genetic foundation of methylation, plus its role in determining an individual's health susceptibility.
(4) Environmental Impact Studies: External environmental factors such as nutrition, toxins, drugs, etc., can potentially impact DNA methylation. WGBS assists researchers in evaluating how these environmental forces might modify gene expression via methylation, thereby influencing an individual's physiological functionality and disease risk.
(5) Evolutionary Research: Furthermore, WGBS can be used to compare DNA methylation patterns amongst different species, shedding light on the role of methylation in evolution. This can contribute to our understanding of how methylation contributes to species adaptation and diversity generation.
Difference Between WGBS and RRBS
WGBS is a high-throughput sequencing technology used for DNA methylation analysis. It can perform methylation analysis on the entire genome, covering each cytosine base and identifying its methylation status, making WGBS the gold standard for DNA methylation research, capable of providing high-resolution and in-depth methylation information. Reduced Representation Bisulfite Sequencing (RRBS) is a 'reduced representation' methylation sequencing method that selectively sequences specific areas in the genome rich in CpG islands and other high methylation regions, as opposed to WGBS. Despite RRBS having a narrower coverage, it is more cost-effective and suited for large-scale sample studies as it requires less sequencing depth.
An in-depth comparative analysis between Whole Genome Bisulfite Sequencing (WGBS) and Reduced Representation Bisulfite Sequencing (RRBS) can elucidate their respective strengths and limitations, assisting researchers in selecting the optimum approach that aligns with their investigative objectives. For instance, should a comprehensive understanding of the methylation status of every cytosine base within the genome be required in the study, WGBS might be a superior choice. Conversely, if the research emphasizes specific methylation regions or necessitates the processing of a large number of samples, RRBS could potentially provide a more cost-effective solution. Moreover, juxtaposing these two techniques can contribute to a better evaluation of their performance and applicability. By contrasting WGBS and RRBS in terms of data volume, cost, coverage, resolution, etc., researchers can garner a richer understanding of the advantages and drawbacks inherent in each method. In turn, this knowledge can guide experimental design and data analysis in a more insightful manner.
Table 4. Difference between WGBS and RRBS
| Feature | WGBS | RRBS |
| Target Coverage | Analyzes the entire genome, covering every C base to determine its methylation status | Adopts a "reduced representation" approach, selectively sequencing specific regions rich in CpG islands and other highly methylated areas, offering narrower coverage but being more cost-effective and suitable for large-scale sample analyses |
| Data Volume and Cost | Generates larger data volumes, hence higher costs | Produces relatively smaller data volumes, leading to lower costs |
| Resolution and Depth of Coverage | Provides higher resolution and deeper coverage, capable of detecting the methylation status of every C base in the genome | Offers comparable high resolution and coverage depth, sufficient to detect the methylation status of selected regions |
| Sample Handling and Experimental Design | Requires more starting DNA material, not suitable for low-input samples or precious clinical samples | Requires less starting DNA material, suitable for low-input sample analyses and large-scale sample studies |
References:
- Fraga, M. F., Esteller, M. (2002). Dna methylation: a profile of methods and applications. Biotechniques, 33(3), 636-49.
- Green, R. E., Krause, J., Briggs, A. W., Maricic, T., Stenzel, U., et al. (2010). A Draft Sequence of the Neandertal Genome. Science, 328(5979), 710–722.
- Hayatsu, H., Negishi, K., & Shiraishi, M. (2004). DNA methylation analysis: speedup of bisulfite-mediated deamination of cytosine in the genomic sequencing procedure. Proceedings of the Japan Academy,80(4), 189-194.
- Herman, J. G., Graff, J. R., Myöhänen, S., Nelkin, B. D., & Baylin, S. B. (1996). Methylation-specific pcr: a novel pcr assay for methylation status of cpg islands. Proceedings of the National Academy of Sciences of the United States of America, 93(18), 9821-9826.
- Ji, L., Sasaki, T., Sun, X., Ma, P., Lewis, Z. A., & Schmitz, R. J. (2014). Methylated dna is over-represented in whole-genome bisulfite sequencing data. Front Genet, 5(5), 341.
- Khanna, A., Czyz, A., & Syed, F. (2013). Epignome[trade] methyl-seq kit: a novel post-bisulfite conversion library prep method for methylation analysis. Nature Methods, 10(10).
- Laird, P. W. (2003). The power and the promise of DNA methylation markers. Nature Reviews Cancer, 3(4), 253–266. doi:10.1038/nrc1045
- Laura-Jayne, G., Mark, Q. T., Lisa, O., Jonathan, P., Neil, H., & Anthony, H. (2015). A genome-wide survey of dna methylation in hexaploid wheat. Genome Biology, 16(1), 273.
- Lin Liu, Ni Hu, Bo Wang, Minfeng Chen, Juan Wang, & Zhijian Tian, et al. (2011). A brief utilization report on the illumina hiseq 2000 sequencer. Mycology, 2(3), 169-191.
- Meissner, A., Gnirke, A., Bell, G. W., Ramsahoye, B., Lander, E. S., & Jaenisch, R. (2005). Reduced representation bisulfite sequencing for comparative high-resolution dna methylation analysis. Nucleic Acids Research, 33(18), 5868-77.
- Meyer, M., Kircher, M., Gansauge, M. T., Li, H., Racimo, F., & Mallick, S., et al. (2012). A high coverage genome sequence from an archaic denisovan individual. Science, 338(6104), 222-6.
- Olova, N., Krueger, F., Andrews, S., Oxley, D., Berrens, R. V., & Branco, M. R., et al. (2018). Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting dna methylation data. Genome Biology, 19(1), 33.
- Raine, A., Manlig, E., Wahlberg, P., Syvänen, A. C., & Nordlund, J. (2016). Splinted ligation adapter tagging (splat), a novel library preparation method for whole genome bisulphite sequencing. Nucleic Acids Research, 45(6), e36.
- Ziller, M. J., Müller, F., Liao, J., Zhang, Y., Gu, H., & Bock, C., et al. (2011). Genomic distribution and inter-sample variation of non-cpg methylation across human cell types. Plos Genetics, 7(12), e1002389.


