Our planet is run by a hidden world of microorganisms. These bacteria, fungi, and other microscopic life forms drive global nutrient cycles, support plant life, and play a key role in our own health. For a long time, scientists could only study the tiny fraction of microbes that could be grown in a lab, leaving the vast majority as a mystery.
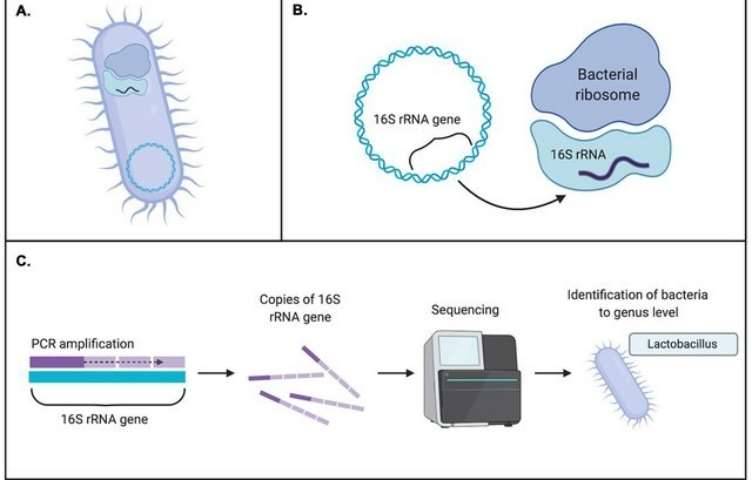
That changed with the development of DNA sequencing methods that don't require growing microbes. Scientists began targeting specific "marker genes"—stretches of DNA that act like a barcode to identify different organisms. This technique, called amplicon sequencing, opened up a new window into the microbial world.
Early methods, however, could only read a short piece of the DNA barcode. This made it difficult to tell closely related species apart, a problem known as low taxonomic resolution. Today, we are in a new era thanks to long-read sequencing. This technology allows us to read the entire, full-length barcode, providing a much more detailed and accurate picture of microbial communities. This resource explores how full-length sequencing of the three main marker genes-16S, 18S, and ITS-is transforming the field of microbial ecology.
Full-length 16S rRNA Sequencing: A High-Resolution Look at Bacteria
The 16S Gene: A Universal Barcode for Bacteria
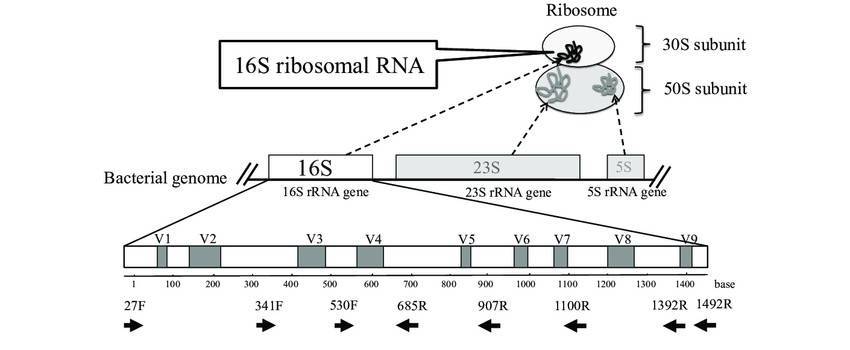
The 16S rRNA gene is the universal standard for identifying prokaryotes (Bacteria and their relatives, Archaea). This gene, approximately 1,500 base pairs (bp) in length, is a crucial part of the ribosome, the cell's protein-making machinery. Its DNA sequence is a mosaic of highly conserved regions, which change very slowly over time and show deep evolutionary relationships, and nine hypervariable regions (V1-V9). These variable regions accumulate mutations more rapidly, creating unique signatures that allow scientists to distinguish between different genera and species, making the gene a perfect barcode for community profiling.
The Limits of Short-Read Sequencing
Relying on short fragments of the 16S gene, such as the V4 region, has been a major limitation. It's like trying to identify a car by only looking at its tire—you miss too many details. This lack of resolution is a significant problem. For instance, a 2024 study of microbes in the Arctic Ocean found that with short-read data, fewer than 10% of the unique DNA sequences found could be identified down to the species level. This prevents scientists from accurately linking specific bacteria to their roles in the environment.
The Technology for a Clearer Picture
Two major technological advances now allow scientists to get accurate, full-length 16S sequences:
High-Fidelity (HiFi) Sequencing: The PacBio sequencing platform uses this method, which reads a single DNA molecule over and over again. This process corrects its own errors, resulting in a long and extremely accurate final sequence.
Optimized Nanopore Sequencing: The Oxford Nanopore (ONT) platform is known for producing very long reads. To improve its accuracy, scientists developed workflows that assign unique sequence labels to each original DNA molecule before amplification and sequencing. These labels help computational tools distinguish true genetic variants from errors introduced during processing, resulting in highly accurate final readouts.
 Figure 1. 16S rRNA gene sequencing analysis performed with MinION™ nanopore technology.( Matsuo, Y. et al, 2021)
Figure 1. 16S rRNA gene sequencing analysis performed with MinION™ nanopore technology.( Matsuo, Y. et al, 2021)
Critical Methodological Bottlenecks
Even with perfect technology, researchers must account for two persistent challenges that can affect their results.
Primer Bias: To sequence a gene, scientists use short DNA strands called "primers" to find and copy the target gene. However, these primers don't work equally well for all microbes. Some microbes' DNA gets copied many times, while others with a slight mismatch might be copied less or missed entirely. Foundational research showed that this bias can cause scientists to miss over 10% of the microbial diversity in a sample.
Database Dependency: A DNA sequence is only useful if you can identify what it belongs to. This requires a large reference database—a digital library of known DNA barcodes. If a microbe is new or not included in the database, it can't be identified. A recent study showed that using a modern, well-curated database called GTDB allowed for far more species-level identifications than the older SILVA database, proving the database is just as critical as the sequencing machine.
Ecological Application Examples
Environmental Health: With precise identification, scientists can now track specific bacterial species involved in cleaning up pollution or causing harmful algal blooms.
Smarter Agriculture: Full-length sequencing allows farmers and researchers to identify the exact soil microbes that are helping or harming their crops, leading to better agricultural practices.
Understanding Human Health: This technology enables scientists to distinguish between beneficial and pathogenic strains of bacteria in the human gut, helping us understand the connection between our microbiome and our health.
Full-length 18S rRNA Sequencing: A Wider View of Microbial Life
The 18S Gene: A Barcode for Tiny Eukaryotes
The 18S rRNA gene is the primary barcode used to identify microscopic eukaryotes. This diverse group includes single-celled protists, algae, amoebas, and other tiny organisms that form the foundation of aquatic and soil food webs. The 18S gene is the eukaryotic counterpart to the prokaryotic 16S gene; it serves the same essential function in the ribosome but is slightly longer, typically ranging from 1,500 to 2,000 bp. This additional length accommodates a more complex structure, but its mix of conserved and variable regions likewise makes it an excellent tool for studying the diversity and evolutionary relationships of microbial eukaryotes.
The Critical Challenge of Primer Choice
For the incredibly diverse world of eukaryotes, the problem of primer bias is even more serious. The specific primers a scientist chooses can dramatically change the results.
A revealing study on amoeba communities in fishponds compared four different 18S primer sets. The results were striking:
No single primer was able to detect all the different types of amoebas in the water.
More alarmingly, no single primer could find all of the potentially disease-causing amoebas that were present.
This shows that a researcher's choice of tool can directly affect their scientific conclusions and even their ability to spot potential health risks.
Other Hurdles: Complex DNA and Incomplete Databases
Eukaryotic DNA presents other challenges. Some organisms have many copies of the 18S gene, which can make them seem more abundant than they really are. Others have extra, non-coding pieces of DNA called "introns" that can interfere with the sequencing process. Furthermore, the reference databases for many protist groups are still incomplete, making identification difficult.
Modern Strategies for Better Results
A Combined Approach: To overcome these issues, scientists now recommend using a multi-primer strategy. This often involves combining full-length sequencing, which gives an accurate identification of the most common species, with a reliable short-read primer that is better at detecting rare organisms.
Real-Time Monitoring for Water Safety: The ability of Nanopore sequencers to produce data in real-time is a major benefit. This allows for the rapid detection of Harmful Algal Blooms (HABs) in lakes and oceans, giving authorities time to issue public safety warnings.
 Figure 2. Field data showing relative ASV read proportions of full-length 18S (left), V4 (center), and V8-V9 (right) rDNA sequences across major protist groups.( Gaonkar CC, P. et al, 2024)
Figure 2. Field data showing relative ASV read proportions of full-length 18S (left), V4 (center), and V8-V9 (right) rDNA sequences across major protist groups.( Gaonkar CC, P. et al, 2024)
Full-length ITS Sequencing: The Gold Standard for Fungi
The ITS Region: The Best Barcode for Fungi
The Internal Transcribed Spacer (ITS) region is the official DNA barcode for identifying fungi and fungus-like organisms. Unlike the 16S and 18S genes, the ITS region is not part of the final ribosome; it is a non-coding spacer found between the 18S and 28S rRNA genes. This region, typically 400-900 bp long, is composed of two subregions, ITS1 and ITS2, separated by the small 5.8S rRNA gene. Because it is not a functional gene itself, the ITS region evolves much more quickly. This rapid evolution results in significant variation between species while remaining consistent within a single species, making it a superior tool for distinguishing between closely related fungi where the more conserved 18S gene might not provide enough detail.
 Figure 3. Fungal rRNA operon targeted amplicon sequencing.( Van Loo, P. et al, 2023)
Figure 3. Fungal rRNA operon targeted amplicon sequencing.( Van Loo, P. et al, 2023)
Why the Full Barcode Is Essential
For many fungi, especially those that cause disease, reading only a small piece of the ITS barcode isn't enough to be certain of their identity.
A key study focused on Phytophthora, a type of fungus-like microbe that destroys agricultural crops. Researchers found these microbes in irrigation water, but they needed a precise way to tell the dangerous species from their harmless cousins. Their analysis showed that only the full-length ITS sequence provided enough detail to reliably distinguish between these critical species. This accuracy is essential for assessing the risk to nearby farms.
What This Means for Health and a Healthy Planet
Protecting Our Food Supply: Full-length ITS sequencing is now a vital tool for agricultural biosecurity. It allows scientists to detect dangerous plant pathogens in soil and water before they can cause widespread damage.
Understanding Ecosystems: This technique helps scientists study the fungi that are essential for forest health, from the symbiotic mycorrhizal fungi that help trees absorb nutrients to the decomposer fungi that recycle dead wood.
Ensuring Food Safety: ITS sequencing is used in the food industry to detect harmful molds that could spoil food or produce toxins, as well as to monitor the helpful yeasts used in making bread, beer, and cheese.
Clinical Diagnostics and Human Health: Long-read ITS sequencing is a powerful emerging tool for rapid clinical diagnostics. For instance, using Nanopore sequencing quickly and accurately identified the pathogen Candida albicans from a pneumonia patient's sputum, matching the results of slower, traditional methods. This shows how scientists are refining the "gold standard" to better distinguish between clinically important fungi.
 Figure 4. Taxonomic classification of fungal mock community across five sequencing regions.( Van Loo, P. et al, 2023)
Figure 4. Taxonomic classification of fungal mock community across five sequencing regions.( Van Loo, P. et al, 2023)
Conclusion
Full-length sequencing of the 16S, 18S, and ITS genes marks a significant step forward in our ability to study the microbial world. It provides a level of detail that was previously impossible, allowing us to move from genus-level guesses to confident species-level identifications.
Yet, as we have seen, each tool requires a thoughtful approach. For 16S, success depends on combining high-accuracy sequencing with modern, complete databases. For 18S, researchers must carefully navigate the major challenge of primer bias, often using multiple strategies to get a complete picture. For ITS, the full-length barcode is proving essential for applications where telling species apart has real-world consequences, like in agriculture and food safety.
The future of microbial ecology lies in integrating these approaches. By combining the information from all three barcodes, and pairing it with other methods that reveal what genes microbes have (metagenomics) or what they are actively doing (transcriptomics), scientists can build a truly comprehensive understanding of the hidden world that supports us all.
References
- Matsuo, Y., Komiya, S., Yasumizu, Y. et al. Full-length 16S rRNA gene amplicon analysis of human gut microbiota using MinION™ nanopore sequencing confers species-level resolution. BMC Microbiol. 2021 Jan 26;21(1):35. https://doi.org/10.1186/s12866-021-02094-5
- Gaonkar CC, Campbell L. A full-length 18S ribosomal DNA metabarcoding approach for determining protist community diversity using Nanopore sequencing. Ecol Evol. 2024 Apr 10;14(4):e11232. https://doi.org/10.1002/ece3.11232
- Ohta A, Nishi K, Hirota K, Matsuo Y. Using nanopore sequencing to identify fungi from clinical samples with high phylogenetic resolution. Sci Rep. 2023 Jun 16;13(1):9785. https://doi.org/10.1038/s41598-023-37016-0