Background
The microbial communities within fermentation pits are crucial determinants of the quantity and quality of light-flavor Baijiu. Typically, genetic diversity and the potential functions of these microbial communities are analyzed using DNA genomics sequencing. However, the characterization of active microbial communities has not been systematically explored. In this study, we employ metatranscriptomic analysis to elucidate the composition, driving factors, and roles of active microorganisms during the fermentation process of light-flavor Baijiu.
Materials & Methods
Sample preparation:
-
Distillery
- RNA extraction
Method:
-
cDNA library construction
- Metatranscriptomic sequencing
- IlluminaHiseq 4000
Data Analysis:
-
De novo assembly
- Functional annotation
- Statistical analysis
Results
Metatranscriptomic sequencing produced 387.99 Gbp of raw data from 2,751,785,770 reads, with 377.27 Gbp of clean data used for analysis. The sequencing had high accuracy, with Q20 values exceeding 98.21%. Assembly results showed contig lengths ranging from 5,686 to 54,864 bp, and unigene numbers averaged 36,834 with lengths of 1,304 bp. The predominant active microorganisms were identified, with 421 genera annotated. The top 20 genera accounted for over 95% of the community, showing significant shifts during fermentation stages.
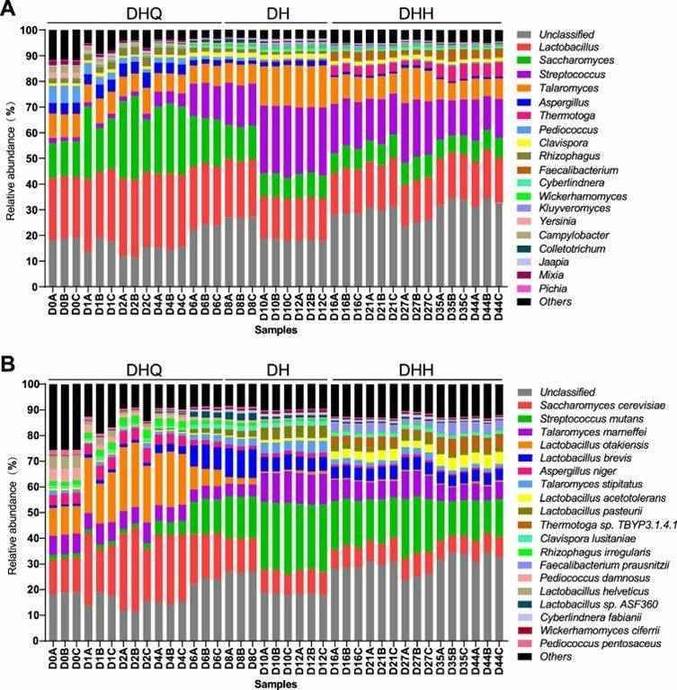 Figure 1. Composition of the active microbial community in light-flavor liquor fermentation.
Figure 1. Composition of the active microbial community in light-flavor liquor fermentation.
Environmental factors such as pH, temperature, and ethanol production influenced microbial succession. Redundancy analysis showed that pH, ethanol, moisture, and starch were key drivers of microbial changes.
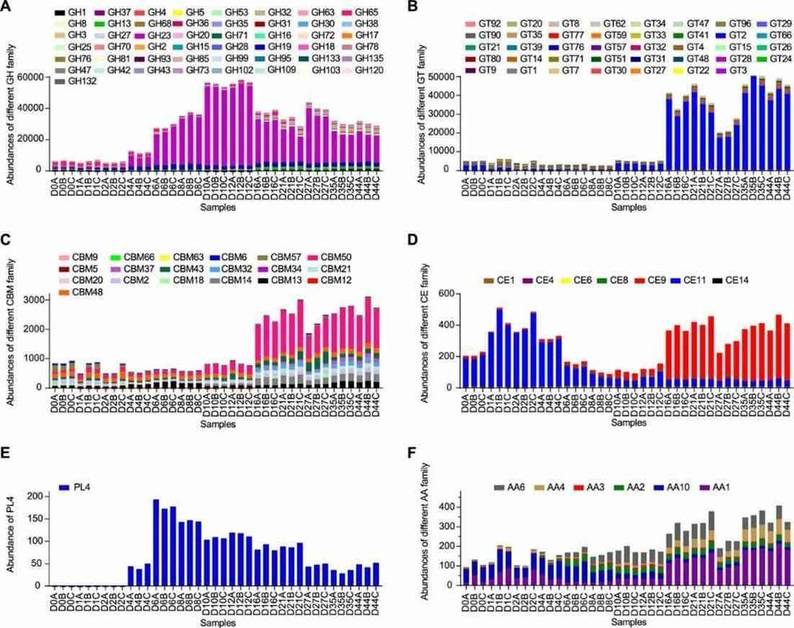 Figure 2. Abundances of the different carbohydrate-active enzyme families in light-flavor liquor fermentation.
Figure 2. Abundances of the different carbohydrate-active enzyme families in light-flavor liquor fermentation.
Carbohydrate-active enzymes (CAZy) showed varying abundances, with glycoside hydrolases (GH) and glycosyltransferases (GT) being the most prominent.
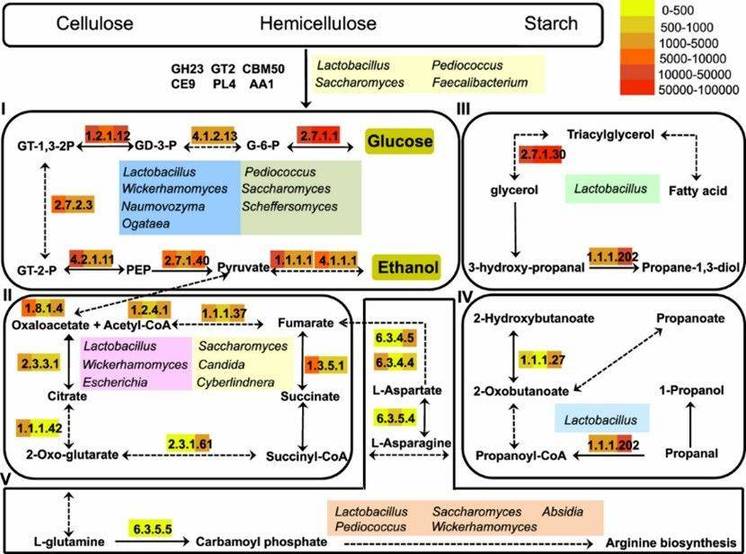 Figure 3. Functional model of carbohydrate hydrolysis, ethanol production, and flavor generation in light-flavor liquor fermentation.
Figure 3. Functional model of carbohydrate hydrolysis, ethanol production, and flavor generation in light-flavor liquor fermentation.
Conclusions
This study used metatranscriptomics to identify active microbes in LFL fermentation, finding Faecalibacterium as a major but poorly understood player. It revealed key microbes responsible for flavor compounds and emphasized the need for improved sampling and fungal genomic resources.