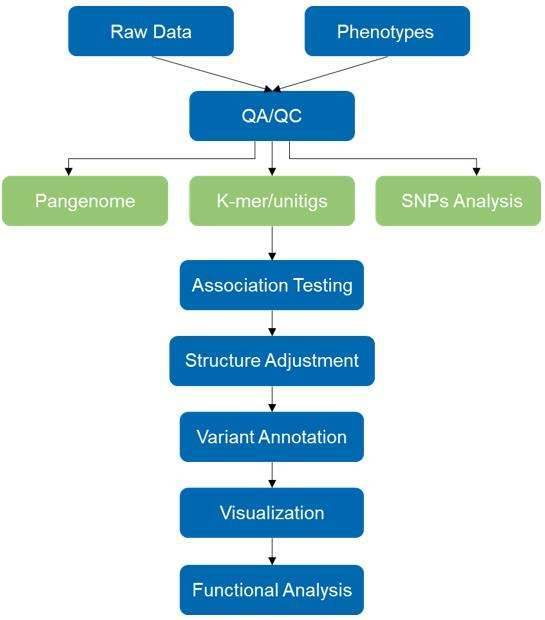
End-to-end solution for your microbial GWAS research
The microbiome has emerged as an important non-genetic component influencing host phenotype and its impact on the host may go beyond the analysis of host genetics. Microbial Genome-Wide Association Studies (mGWAS) allows revealing the characteristics and mutations of microbiota associated with the host. mGWAS utilizes and develops human GWAS research methods to resolve the interactions and associations between variation (such as single nucleotide polymorphisms (SNP) and INDEL, gene presence-absence, copy number variation (CNV) and sequence inversions (SI)) and phenotypes in the microbial genome with the host and environment.
Our mGWAS service offers many great benefits and conveniences, such as a high-throughput bioinformatics pipeline and established reference database, as well as providing sequencing control at a cost per sample. The use of this strategy demonstrates a significantly different characterization of the microbiota and greatly deepens our understanding of its role in various body regions of human health and disease.