Background
Cancer immunotherapy with CAR-T cells often faces challenges with T cell persistence and efficacy. The EpiVIA assay addresses this by simultaneously mapping lentiviral integration sites and chromatin accessibility at cellular resolution. It uses Tn5 transposase to insert sequencing adapters into viral-host DNA fragments, revealing integration sites with high accuracy. This method shows CAR-T integrations favoring specific genomic regions and provides insights into T cell behavior and treatment durability.
Materials & Methods
Method:
-
ATAC-Seq
- Integration site analysis
Data Analysis:
-
Identification of integration site
- Local chromatin state of integration sites
- Combined analysis of host chromatin state, integration site, and provirus accessibility.
Results
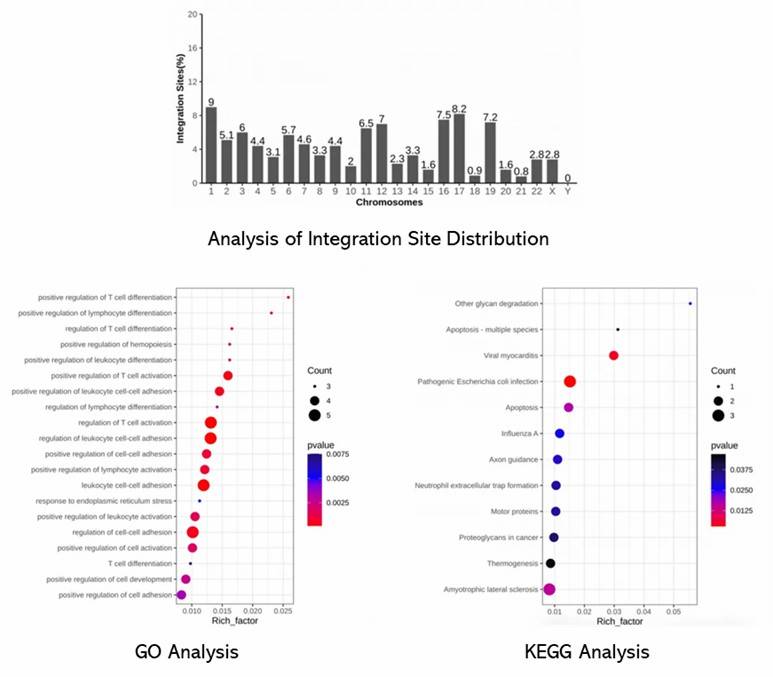
EpiVIA detects CAR-T integration sites at the individual cell level effectively. In a study of 5,000 CD8+ T cells, it identified 193 unique integration sites. Higher sequencing depth improved detection sensitivity, with more fragments per cell enhancing the ability to identify integration events.
EpiVIA's analysis of CAR-T integration sites reveals that 60% occur in transposable elements, predominantly Alu repeats, and are enriched in intronic regions. Integration sites identified by EpiVIA closely align with known HIV-1 integration sites and recurrent integration genes (RIGs). The analysis confirms EpiVIA's accuracy and highlights its ability to detect integration hotspots at an individual cell resolution.
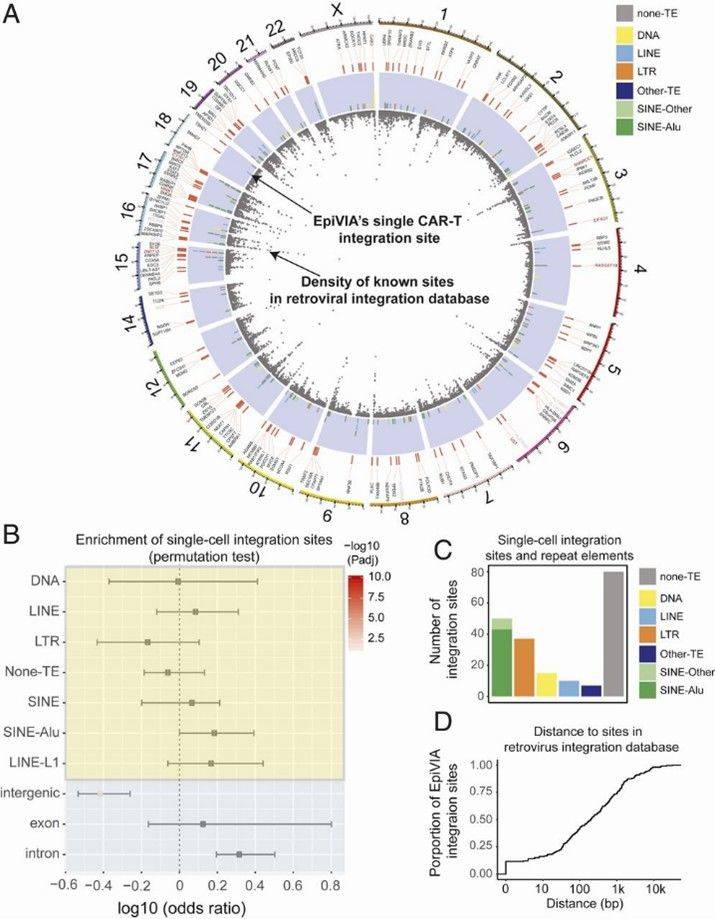 Fig 1. Genomic features of EpiVIA identified integration sites in individual cells.
Fig 1. Genomic features of EpiVIA identified integration sites in individual cells.
Conclusions
EpiVIA is a tool for profiling both epigenomes and lentiviral integration sites at bulk and cellular resolution. It accurately maps integration sites, cell fate, and chromatin accessibility, overcoming limitations of existing methods.

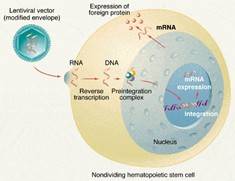
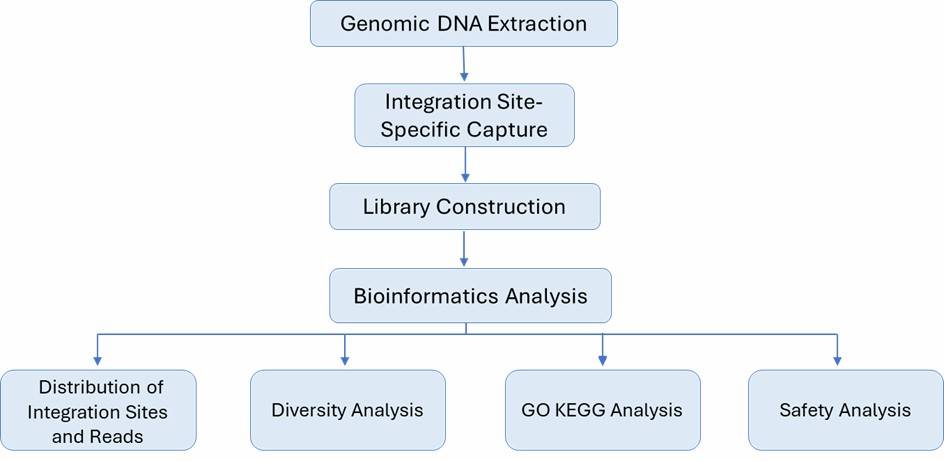
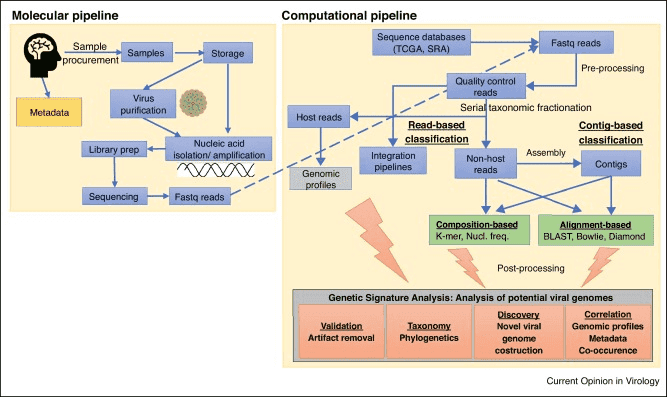
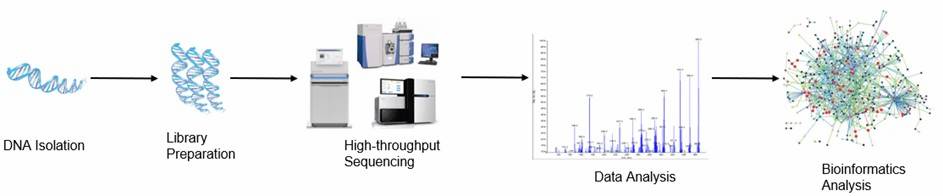 Figure 1. The high-throughput sequencing analysis process.
Figure 1. The high-throughput sequencing analysis process.