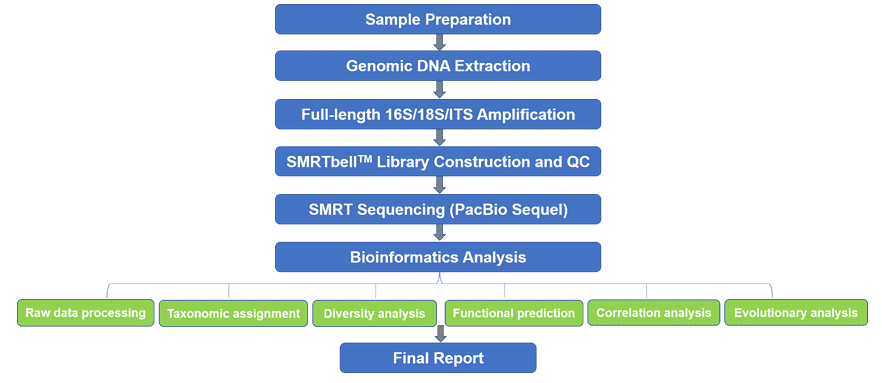
The 16S rRNA, 18S rRNA, and internally transcribed spacer (ITS) are commonly used molecular markers for species identification and phylogenetic analysis in prokaryotes or eukaryotes. While 16S and 18S rRNAs contain several hypervariable regions that vary dramatically in length, ITS tends to be hypervariable and unique among fungal species, even strains. These hypervariable sequences can be used to identify and characterize microbial diversity, function, and evolution analysis. The PacBio SMRT technology is a long-read sequencing platform with an average read length of about 10~18 Kb, which can cover all variable regions of 16S/18S/ITS. Our SMRT-based 16S/18S/ITS sequencing service can accurately amplify the full-length 16S/18S/ITS with validated universal primer pairs that target the highly conserved regions flanking these sequences. Compared with short-read sequencing data only from variable regions, full-length 16S/18S/ITS sequencing enables more accurate species/strain classification and identification.
SMRT-based 16S/18S/ITS sequencing enables us to identify or characterize the prokaryotic species and explore microbial diversity in complex microbial communities. In addition, this sequencing platform can advance microbial taxonomy and phylogenetic studies. In the medical field, it can be used to analyze the global microbial communities in both healthy and diseased groups for biomarker discovery, disease diagnosis, and target discovery. SMRT-based 16S/18S/ITS sequencing has also been applied to genetic changes tracking, food sources monitoring, reflection of biomass concentration, marine environmental conservation, surveillance and response to infectious disease outbreaks, and industrial manufacturing.