Backgrounds
Pseudomonas bacteria can survive in harsh environments like crude oil by producing enzymes and rhamnolipids (RLs) that help degrade alkanes. This study compared three such strains, finding they have flexible genomes and high levels of alkane-degrading enzymes, making them suitable for bioremediation and oil recovery.
Methods
Sample preparation:
-
P. aeruginosa strains
- DNA extraction
Data Analysis:
-
Genome assembly
- Annotation
- Phylogenetic analysis
- Comparative genome analysis
Results
IMP66, IMP67, and IMP68 were isolated from crude oil, and their complete genomes were sequenced. Phylogenetic analysis using ten housekeeping genes confirmed that IMP67 and IMP68 are P. aeruginosa, while IMP66 was also identified as P. aeruginosa. A maximum likelihood tree of 54 Pseudomonas strains and core genome analysis of 90 P. aeruginosa isolates revealed that all three strains clustered together in the same group, indicating they are closely related within the P. aeruginosa species.
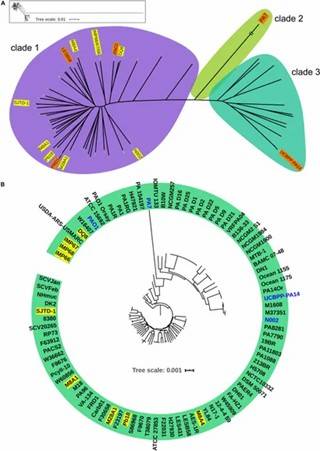 Figure 1. Phylogeny of P. aeruginosa population structure.
Figure 1. Phylogeny of P. aeruginosa population structure.
IMP66, IMP67, and IMP68 have similar genome sizes and GC content, with about 6,100 coding sequences (CDSs) each, which is higher than the well-studied PAO1 strain. Compared to PAO1, most crude oil isolates have larger genomes, potentially aiding their survival. Comparative analysis revealed 592 unique genes in IMP66, IMP67, and IMP68, mostly related to mobile genetic elements and gene functions such as transcription and cell wall biogenesis. Horizontal gene transfer (HGT) appears common in these strains, with conserved regions indicating potential gene acquisition and adaptation advantages, including features linked to alkane degradation.
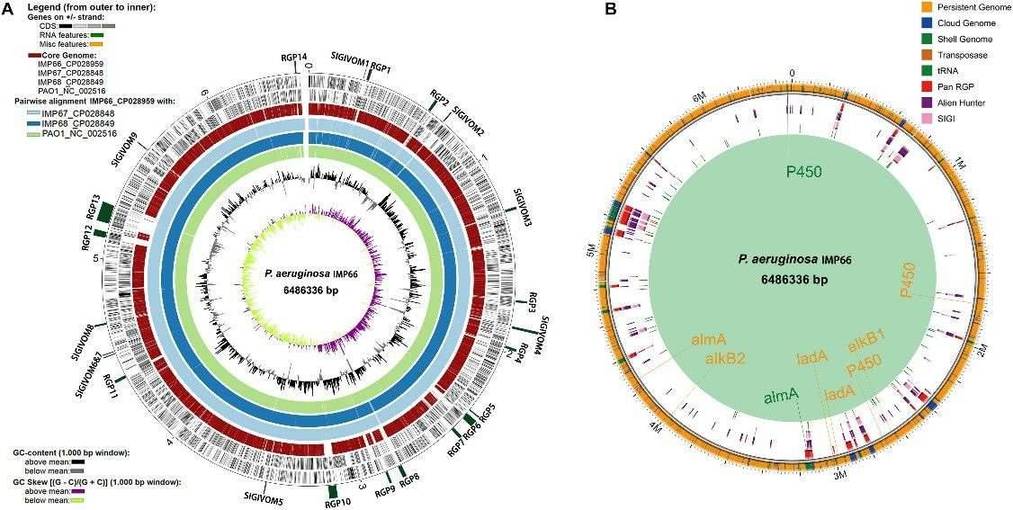 Figure 2. Whole genome comparison of IMP66, IMP67, IMP68, and PAO1.
Figure 2. Whole genome comparison of IMP66, IMP67, IMP68, and PAO1.
Conclusions
P. aeruginosa strains IMP66, IMP67, and IMP68, isolated from crude oil, are highly adaptable, excelling in alkane degradation and rhamnolipid production. They have numerous alkane hydroxylase genes and show signs of horizontal gene transfer, enhancing their survival. Their efficient QS signaling helps balance rhamnolipid production. These traits make them promising for bioremediation and industrial applications, with their CRISPR-Cas system offering potential for genetic engineering.