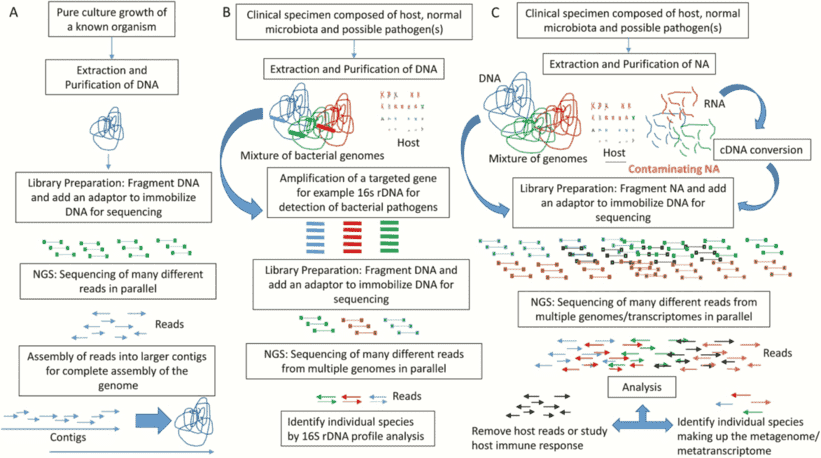
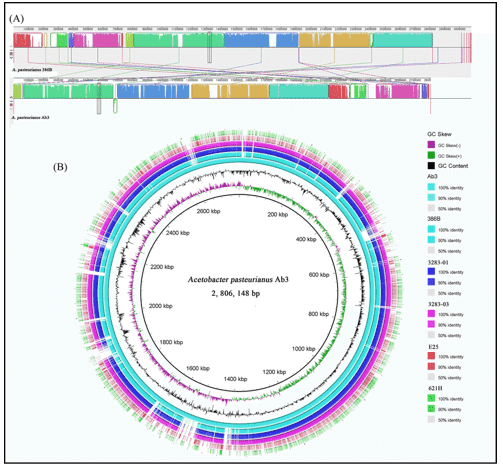
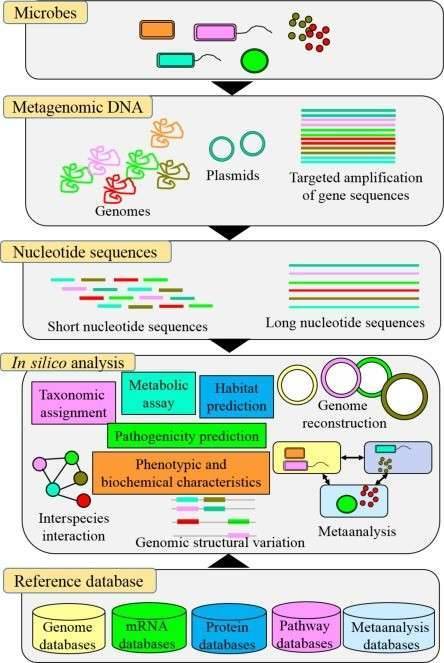
The majority of the biochemical interaction on the planet is carried out by microbial populations and they perform an essential part in bioprocesses such as metabolism and immune homeostasis in the human microbiome. In microbial genomics, the change to high-throughput sequencing technologies has drastically altered our comprehension of microbial populations in various ecosystems. There is now an even more change in the acknowledgment of the intricacy of these populations as more publicly accessible microbiome databases centered on shotgun metagenomic sequencing are becoming accessible. Whole genome sequencing is becoming a progressively viable supplement to clinical research in the prediction of antimicrobial resistance genes (ARGs) and mobile genetic elements (MGEs) in bacterial sequences. And whole genome databases enable specific genes and their variants to be characterized, in addition to setting up the taxonomy and comparable abundance of microbial populations.
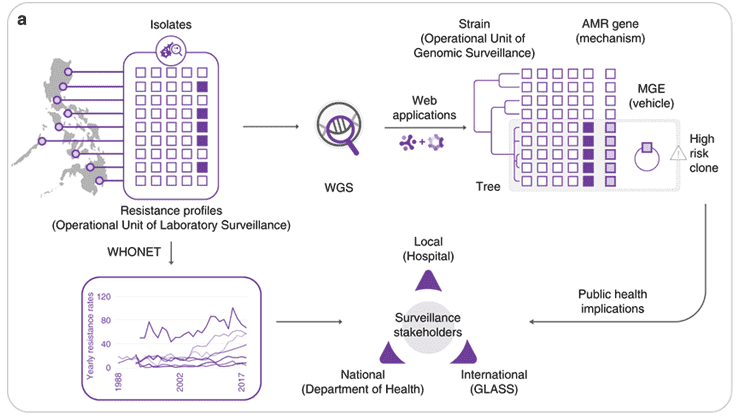 Figure 1. Implementing whole-genome sequencing (WGS) for AMR surveillance.
Figure 1. Implementing whole-genome sequencing (WGS) for AMR surveillance.
Mobile genetic elements (MGEs) are crucial to our knowledge of how genes (and their associated components) move within a community through horizontal gene transfer within a community. The structure of microbial populations, their diversity and density, and their interaction with the environment can have a lasting effect on these components. Thereby, the status of these MGEs (mobilomes) appears to be a major factor in affecting differences in the shape of bacterial diversity and their influence on the host organism or tissue caused by selection pressure. The motion of antimicrobial resistance indicators and virulence factors between microbes is also caused by MGEs.
Ideas on how ARGs travel along all multiple genomes within the microbiome can be provided by profiling the mobilome and its affiliated ARGs. All sequences of MGEs need to be defined from metagenomic sequencing data to characterize the mobilome in bacterial diversity and would preferably be appointed to a microbial host. Although it is a common approach that is significantly simpler to identify MGEs from single isolates using whole-genome sequencing, metagenomic sequencing is progressively being utilized to identify and categorize multiple MGEs from bacterial diversity.
In the pre-WGS era, the assurance of the science and hereditary qualities of MGEs was difficult, time-consuming, and usually constrained by the accessibility of suitable strategies. These days, the accessibility of short- and long-read sequencing methods for WGS profiling sheds light on research into bacterial genomics and gives rich data of the substance and differing qualities of MGEs (such as plasmids, bacteriophages, transposons). For the most part, DNA sequences associated with MGEs of unrelated microorganisms can be effortlessly recognized due to the rich G + C content that differs to some extent from that of their hosts, demonstrating prior events of lateral gene transfer.
Each day, a gigantic number of bacterial genomes are being sequenced utilizing NGS in research facilities over the globe. With this tremendous sum of information accessible, it is vital to extract project-relevant information effortlessly. Be that as it may, most of the bacterial genomes are accessible as contigs that have been built utilizing auto-annotation algorithms in public databases. Over the years, profoundly productive strategies for bacterial genome annotation have been developed that do not need much manual input.
References
- Hua X, Liang Q, Deng M, et al. BacAnt: A Combination Annotation Server for Bacterial DNA Sequences to Identify Antibiotic Resistance Genes, Integrons, and Transposable Elements. bioRxiv. 2021 Jan 1:2020-09.
- Uelze L, Grützke J, Borowiak M, et al. Typing methods based on whole genome sequencing data. One Health Outlook. 2020 Dec;2(1).
- Argimón S, Masim MA, Gayeta JM, et al. Integrating whole-genome sequencing within the National Antimicrobial Resistance Surveillance Program in the Philippines. Nature communications. 2020 Jun 1;11(1).
- Bello-López JM, Cabrero-Martínez OA, Ibáñez-Cervantes G, et al. Horizontal gene transfer and its association with antibiotic resistance in the genus Aeromonas spp. Microorganisms. 2019 Sep;7(9).