Introduction to Our Viral Whole Genome Sequencing Platform
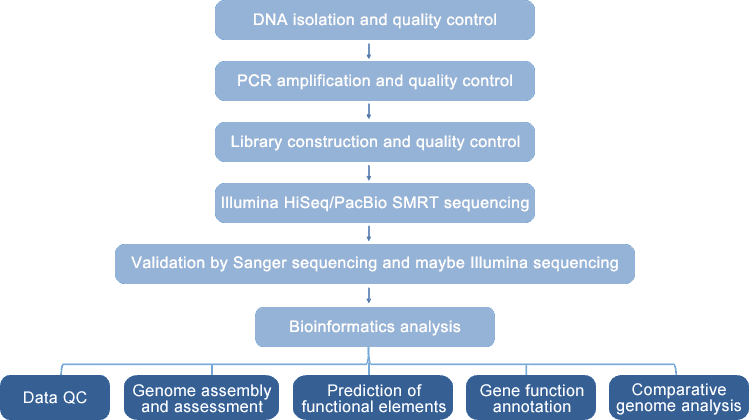
The automated and excellent vrial whole genome sequencing platform at CD Genomics enables us to sequence and assemble complete viral/phage genomes de novo or with the help of the reference genome. We perform PacBio SMRT (10/20 Kb library, 100-150X) and/or Illumina HiSeq PE150 (300-500 bp library, 100-200X) sequencing based on your sample type. PacBio SMRT sequencing can produce long reads, which is advantageous for genome assemblies, especially in GC-rich and repeat-dense regions. And illumina HiSeq sequencing is well-accepted due to its high throughput and accuracy. After sequencing, raw data are processed by quality control, trimming, and filtering. Accurate reads are then used for gap-closed genome assemblies or draft genome assemblies based on your needs.
Based on our viral whole-genome sequencing platform, we offer reliable viral whole-genome resequencing, de novo sequencing, and genome survey sequencing services. The resulting genome assemblies are confidential and ready for synteny analysis, variation analysis, and evolutionary analysis. Viral whole genome sequencing is also a tool for understanding the biochemical interactions between prokaryotic cells and phages, and has been widely applied to food industry and medical research. For example, multi-drug resistance is increasing sharply, and phage therapy is an efficient way to treating bacterial infections. Viral whole genome sequencing can help promote the development and spread of phage therapy in the United States and other countries.