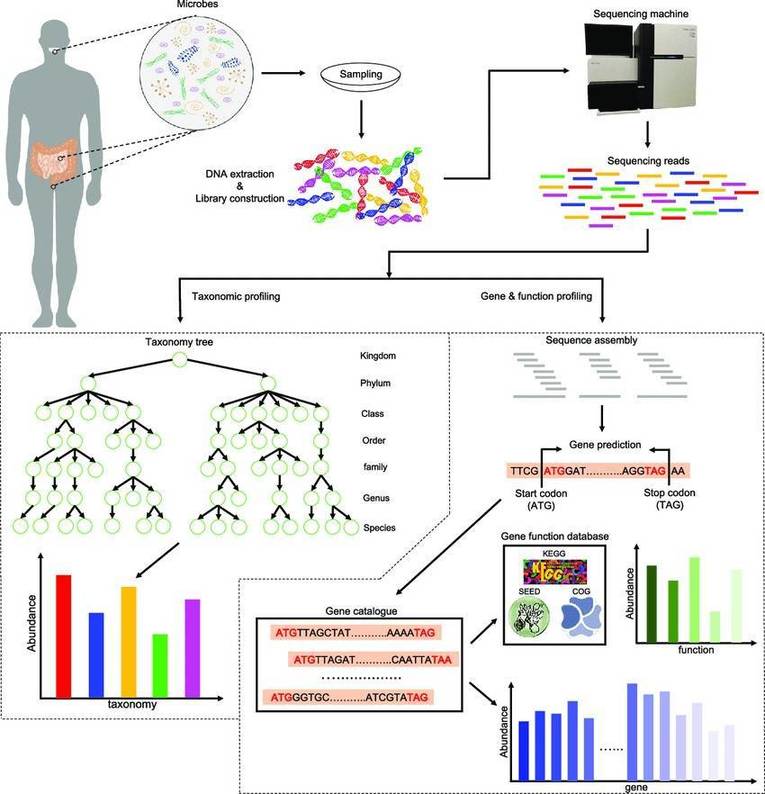
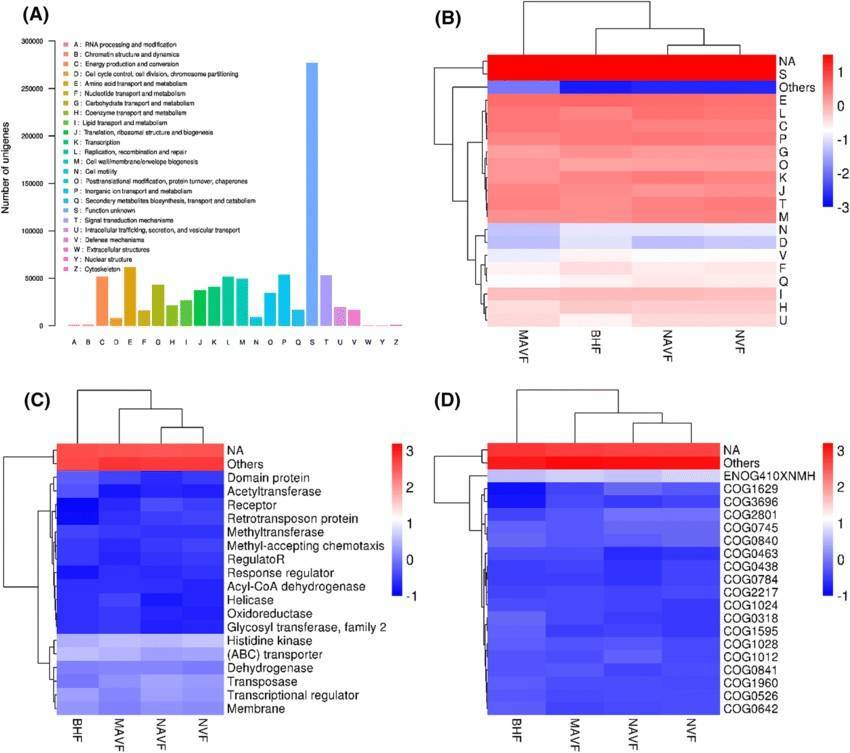
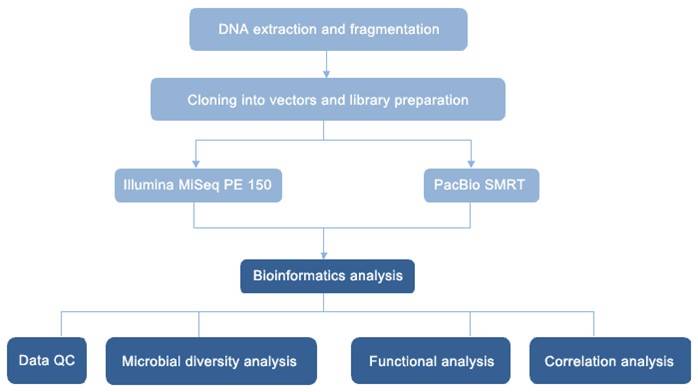
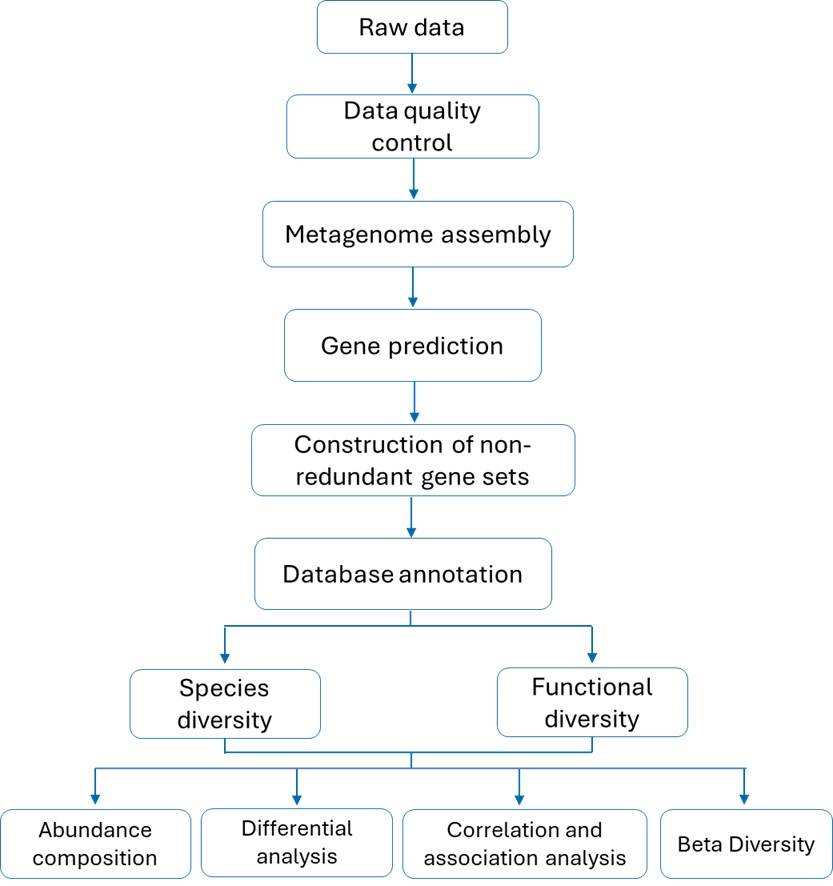
Our metagenomics platform aims to study novel genes, microbial pathway, microbial diversity, evolution, functional annotations, and correlation analysis by utilizing the next/third generation sequencing technology. Our scientists utilize this platform to obtain fast, accurate, and cost-effective results.
Our Advantages:
- High Depth of Coverage: We provide sequencing with high coverage, enabling detailed species identification and functional gene analysis.
- Integrated Functional Analysis: Our services include not just taxonomic profiling but also functional annotation, allowing you to explore metabolic pathways and interactions.
- Diverse Sample Compatibility: We accept a wide range of sample types, including soil, water, and biological tissues, catering to various research fields.
- User-Friendly Data Reports: Results are delivered in clear, actionable formats, making it easy for researchers to interpret and utilize the data in their studies.
What is the Microbial Metagenome
The microbial metagenome represents the complete set of genetic material found in a particular environment, including a variety of microorganisms like bacteria, archaea, fungi, and viruses. Unlike traditional microbiology, which often relies on isolating and culturing individual species, metagenomics utilizes cutting-edge next-generation sequencing (NGS) to analyze DNA extracted directly from environmental samples. This method opens the door for researchers to investigate the complex web of interactions within microbial communities, shedding light on their diversity, functional roles, and contributions to ecosystem dynamics. By mapping out the metagenome, scientists can uncover how these microorganisms influence crucial processes such as nutrient cycling, disease mechanisms, and overall ecosystem health.
Why Perform Microbial Metagenomics
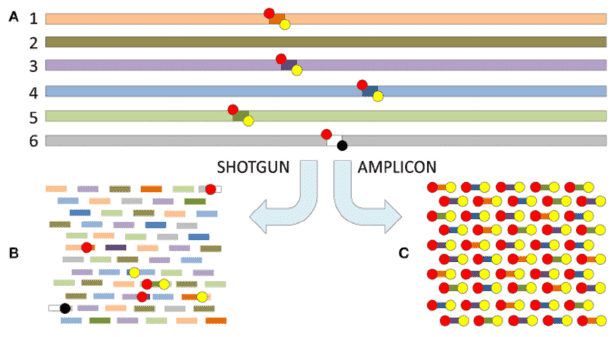
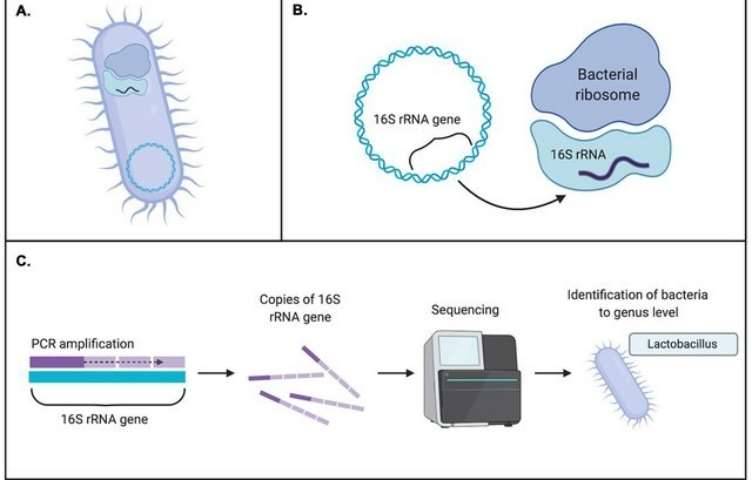
Conducting microbial metagenomics is vital because it allows scientists to sequence DNA from environmental samples directly, bypassing the tedious and often incomplete work of isolating individual strains. Traditional methods, such as 16S rRNA gene sequencing, provide only a limited view of the functional potential within microbial communities. In contrast, metagenomics captures a wider array of genetic information, offering a richer understanding of microbial diversity and their metabolic functions. This information plays a crucial role in various fields, from environmental monitoring to advancements in human health and biotechnology, facilitating improvements in diagnostics, therapeutic approaches, and bioremediation techniques.
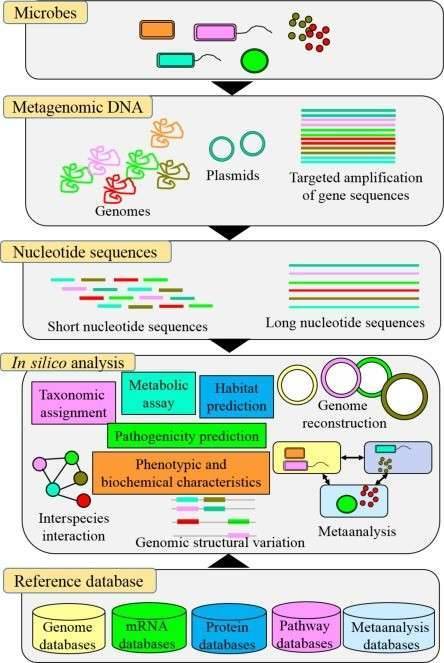
What is the Difference Between Microbial Genomics and Metagenomics
Microbial genomics is primarily concerned with the genetic material of individual microbial species, often requiring the isolation of pure cultures for detailed genome sequencing. On the other hand, metagenomics examines the combined genomic content of entire microbial communities without needing to isolate specific organisms. While microbial genomics provides in-depth insights into the genetics and functions of particular species, metagenomics reveals the intricate relationships and interactions among diverse microorganisms in their natural environments. This distinction is important; metagenomics not only identifies which organisms are present but also explores their functional capabilities, leading to a more comprehensive understanding of microbial ecology.

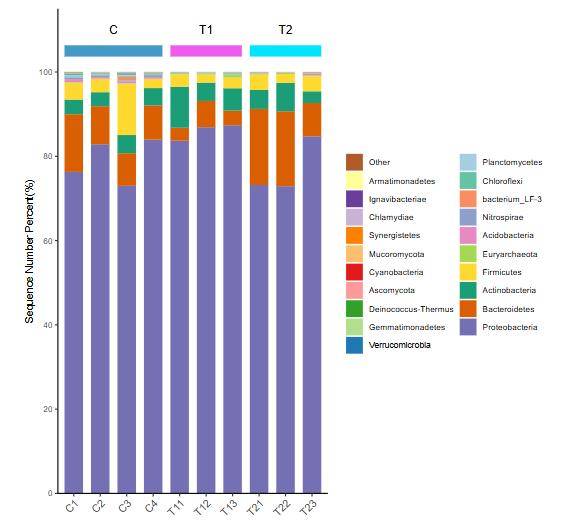 Species Distribution Bar Chart
Species Distribution Bar Chart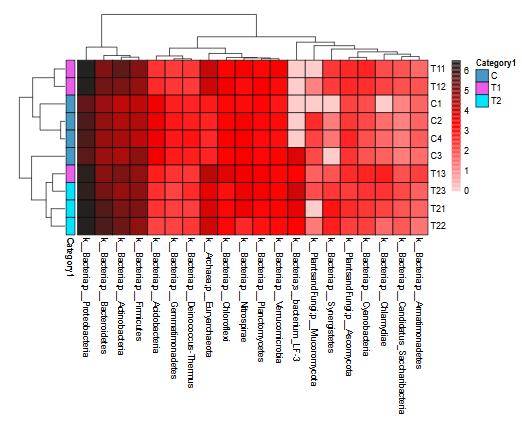 Heatmap
Heatmap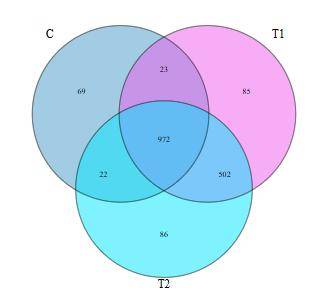 Venn Diagram
Venn Diagram LEfSe Analysis LDA Bar Chart
LEfSe Analysis LDA Bar Chart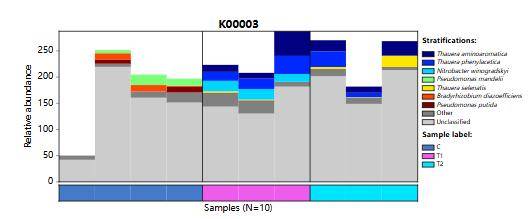 Enzyme Activity Gene Species Source Composition Bar Chart
Enzyme Activity Gene Species Source Composition Bar Chart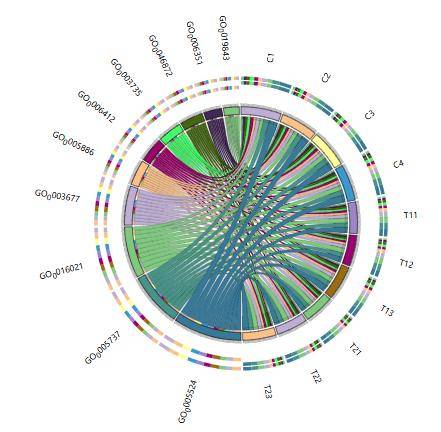 Gene Ontology Distribution Circos Plot in Various Samples
Gene Ontology Distribution Circos Plot in Various Samples
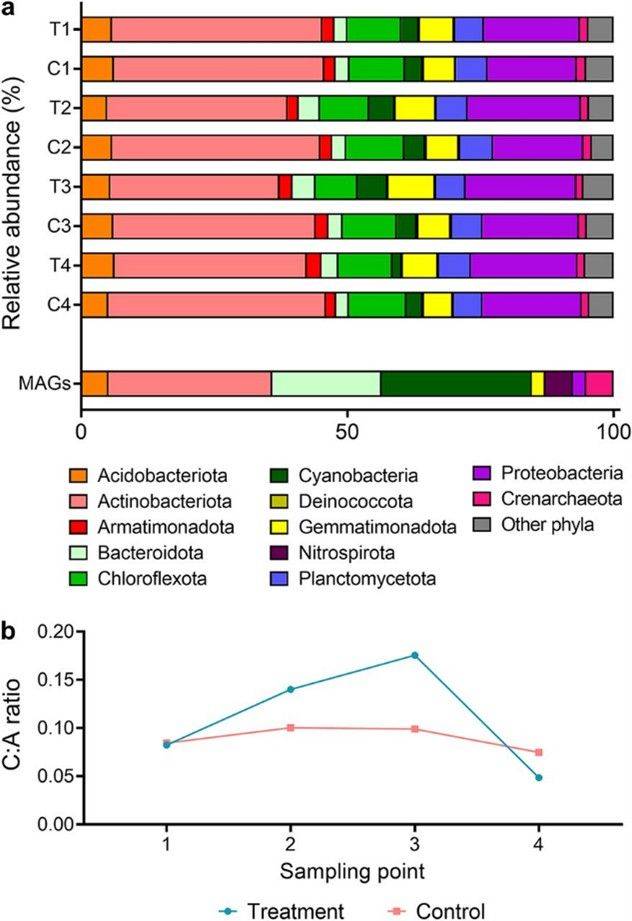 FIG 1 Changes in phylum-level community composition of the Australian desert soil microcosms during hydration and desiccation.
FIG 1 Changes in phylum-level community composition of the Australian desert soil microcosms during hydration and desiccation.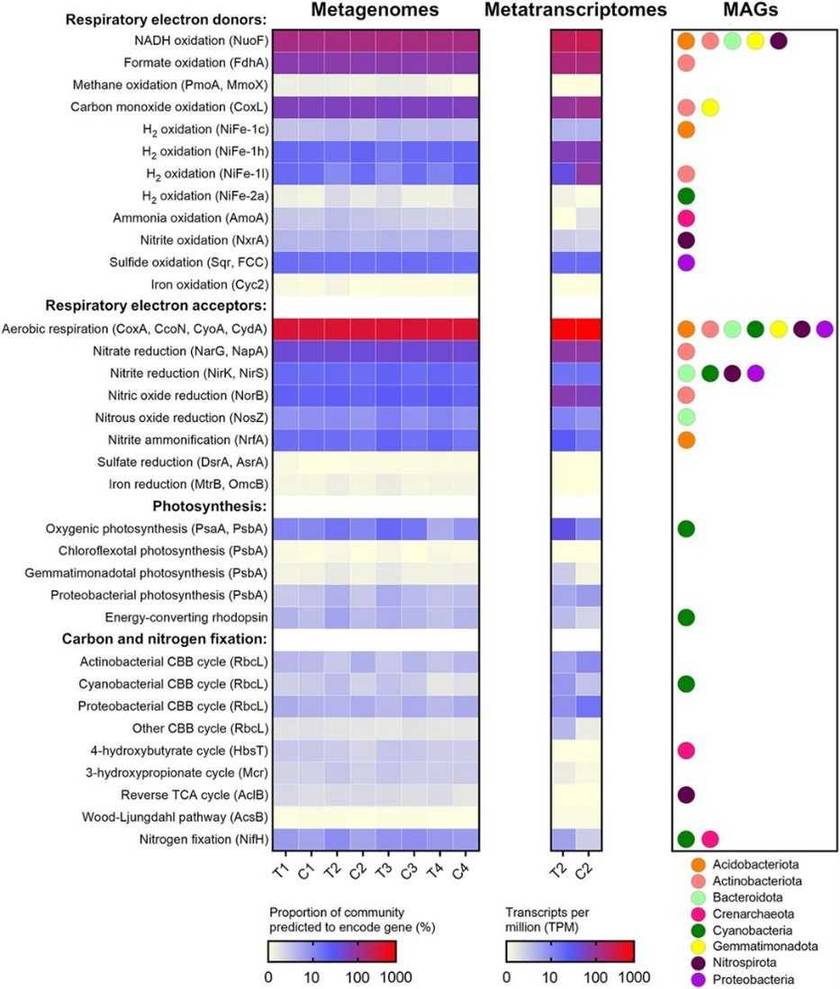 FIG 2 Abundance, expression, and distribution of metabolic marker genes in the Australian desert soil microcosms.
FIG 2 Abundance, expression, and distribution of metabolic marker genes in the Australian desert soil microcosms.