What is 16S rRNA Sequencing
16S rRNA gene sequencing is a kind of amplicon sequencing that targets and reads an area of the 16S rRNA gene, which is found in all Bacteria and Archaea, so it can only define these microorganisms. Other kinds of amplicon sequencing, like ITS sequencing for fungi or 18S sequencing for protists, can distinguish various microorganisms.
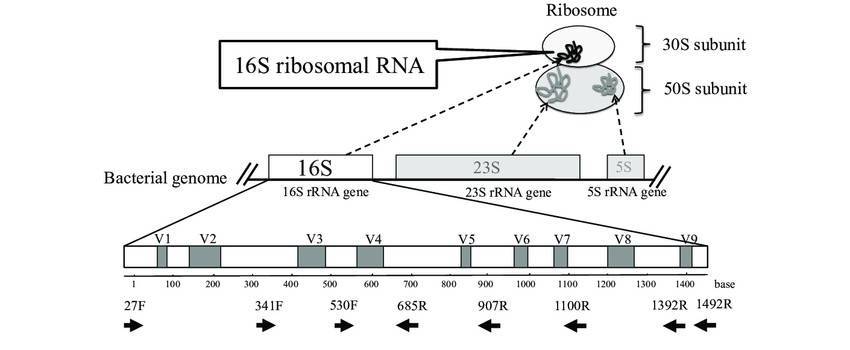
The 16S ribosomal RNA (rRNA) gene encodes a segment of rRNA present universally across bacterial genomes. This gene comprises conserved regions, which elucidate phylogenetic relationships among diverse species, as well as variable regions, which provide insights into interspecies differences. Sequencing of the 16S rRNA gene frequently employs MiSeq technology, specifically targeting the V3-V4 variable regions to facilitate the investigation of microbial diversity. Through the sequencing of this gene, it is possible to identify distinct genera and species within a microbial community and to assess their relative abundances.
This method comprises a series of crucial procedures, commencing with sample acquisition from diverse environments or biological reservoirs, followed by DNA extraction while ensuring the preservation of bacterial DNA integrity. Subsequent to this, the 16S rRNA gene undergoes amplification through polymerase chain reaction (PCR) using primers specifically designed to target conserved regions and amplify variable regions like V3-V4, V4, and V6-V8. The selection of primers holds utmost significance, given that the choice of primers can influence the preferential amplification of distinct bacterial taxa. The amplified 16S rRNA genes are subsequently subject to sequencing using state-of-the-art technologies such as Illumina MiSeq, renowned for its high precision and coverage depth, surpassing preceding techniques like 454 pyrosequencing in both performance and cost-efficiency. Following sequencing, data processing ensues, entailing the removal of low-quality reads, and the trimming of adapters and primers. High-quality sequences are then grouped into Operational Taxonomic Units (OTUs) or Amplicon Sequence Variants (ASVs) based on sequence homology.
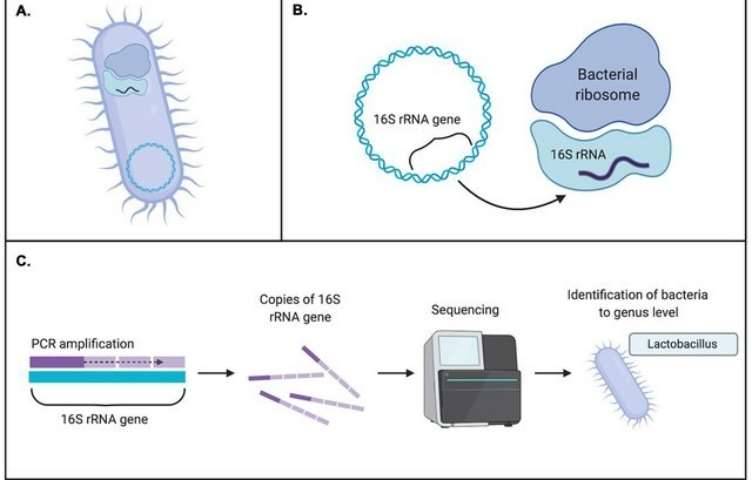 Figure 1. 16S rRNA sequencing is the most common method used to analyze the microbiome. (Patel et al., 2022)
Figure 1. 16S rRNA sequencing is the most common method used to analyze the microbiome. (Patel et al., 2022)
The result of 16S rRNA sequencing is sequencing 'reads' (DNA sequence strings) that can be analyzed using a number of basic bioinformatic processes, which are referred to as 'pipelines' when they are integrated. These bioinformatic pipelines 'clean' the data by removing sequencing errors or questionable reads, which can then be associated with microbial genomic databases to correctly determine and profile the bacteria (and archaea) that were visible in the specimens.
Applications of 16S rRNA Sequencing
Medical Microbiology
The utilization of 16S rRNA sequencing holds significant value in pathogen identification and disease diagnosis within the realm of clinical microbiology. This method enables the identification of bacteria and archaea in clinical specimens, offering a crucial means to detect pathogens that might be challenging to culture through traditional methods. An illustrative example of its effectiveness can be observed in the work of Chiu and Miller (2019) where the technique was successfully employed to diagnose infections by detecting bacterial DNA in clinical samples that had previously eluded diagnosis using conventional approaches.
In a separate investigation, Yan et al. (2021) conducted a study analyzing stool samples from individuals with Parkinson's disease (PD) and healthy controls through a combined approach of 16S rRNA gene sequencing and gas chromatography-mass spectrometry (GC-MS). Their findings revealed an elevated abundance of genera like Alistipes, Rikenellaceae_RC9_gut_group, Bifidobacterium, and Parabacteroides in PD patients, contrasted with a reduction in Faecalibacterium levels. These outcomes suggest novel avenues for potential therapeutic interventions in PD by means of dietary modifications and regulation of the gut microbiota.
Environmental Microbiology
In environmental studies, 16S rRNA sequencing is crucial for analyzing microbial diversity and ecological functions. It helps monitor the impact of pollution and other environmental changes on microbial communities. For example, Delgado-Baquerizo et al. conducted a global study of soil microbiomes in urban greenspaces to compare their diversity and composition with nearby natural ecosystems, identify consistent microbial residents, evaluate influencing socioeconomic and environmental factors, and assess microbial functional attributes. This work advances our understanding of soil organisms' roles in urban environments and supports sustainable urban greenspace development. Researchers analyzed soil microbial communities using ribosomal DNA analysis to understand their global distribution and response to environmental changes. Findings showed high bacterial diversity and stress-induced variations in community composition. This research informs soil conservation and management strategies amidst climate change and enhances our predictive capabilities for microbial community dynamics in changing environments (Dargiri et al., 2023).
3. Agriculture
In agriculture, 16S rRNA sequencing helps understand the soil microbiome and its impact on crop health and yield. For instance, Fan et al. employed high-throughput sequencing to investigate soil microorganisms for biological control of Fusarium wilt in bananas. Soil microbial diversity analysis in Yunnan's banana-producing areas revealed significant enrichment of Bacillus, negatively correlated with the pathogen Foc TR4. Isolation of Bacillus velezensis strain YN1910 from disease-suppressive soils demonstrated significant control (78.43–81.76%) of banana Fusarium wilt and growth promotion in banana plants in both in vitro and pot experiments. Another study by Bulgarelli et al. (2012) explored the root microbiome of Arabidopsis thaliana, offering insights into plant-microbe interactions and their influence on plant health.
4. Industrial Applications
16S rRNA sequencing is a valuable tool in industrial microbiology for optimizing microbial processes utilized in manufacturing. Cao et al. (2016) employed Illumina high-throughput sequencing to investigate microbial communities within an ANAMMOX-UASB reactor treating high-strength wastewater. Remarkably, the reactor reached a stable operational state within a mere 26 days, showcasing a notable nitrogen removal efficiency. The study observed a gradual increase in the abundance of ANAMMOX bacteria over time, with Candidatus Brocadia emerging as the predominant species. The community composition was primarily constituted by Chloroflexi, Bacteroidetes, and Proteobacteria, with the presence of Denitratisoma from the Proteobacteria phylum noted as well. Furthermore, scanning electron microscopy (SEM) images confirmed the presence of a diverse array of microorganisms within the reactor.
In another industrial context within the food industry, researchers conducted an investigation analyzing fermented milk products (FMPs) originating from multiple regions of Russia. Employing 16S rRNA gene sequencing, the study focused on characterizing the bacterial communities present in these products. The results unveiled that Firmicutes, encompassing genera such as Lactococcus, Lactobacillus, Streptococcus, Lentilactobacillus, and Leuconostoc, were the prevailing taxa, followed by Proteobacteria. Notably, the abundance of Lactobacillus and Lactococcus was particularly prominent in FMPs crafted using raw milk and subject to fermentation at room temperature. Conversely, products produced from pasteurized milk and fermented at higher temperatures were predominantly enriched with Streptococcus. The comparative analysis across different FMPs illuminated both shared microbial profiles and distinctive regional specificities, hinting at promising prospects for innovative product development and enhanced characteristics (Kochetkova et al., 2022).
5. Animal Health
In veterinary science, 16S rRNA sequencing investigates the microbiomes of different animal species, shedding light on their roles in nutrition, immune function, and disease resistance. For example, Kormas et al. (2014) compared gut prokaryotic communities in wild, organically-, and conventionally reared sea bream using 16S rRNA gene sequencing. The number of bacterial OTUs decreased from wild to conventionally reared fish. This study highlights diet-induced changes in sea bream gut microbiota. Another study by Suchodolski (2011) focused on the gut microbiota of dogs and cats, providing valuable information for developing probiotics and other microbiome-targeted therapies.
6. Forensic Science
Forensic applications of 16S rRNA sequencing include microbial forensics, where microbial signatures at crime scenes can be used to link suspects to locations or objects. Emmons et al. (2020) studied microbial colonization of human bone and tooth samples to understand postmortem skeletal degradation. They identified dominant bacterial and eukaryotic colonizers, including Proteobacteria and Ascomycota. However, bacterial loading did not significantly correlate with human DNA concentration, indicating complex interactions between microbial communities and DNA preservation. Similarly, Lax et al. (2015) utilized 16S rRNA sequencing to explore the microbial "fingerprints" left on surfaces, aiding forensic identification.
What is Shotgun Metagenomic Sequencing
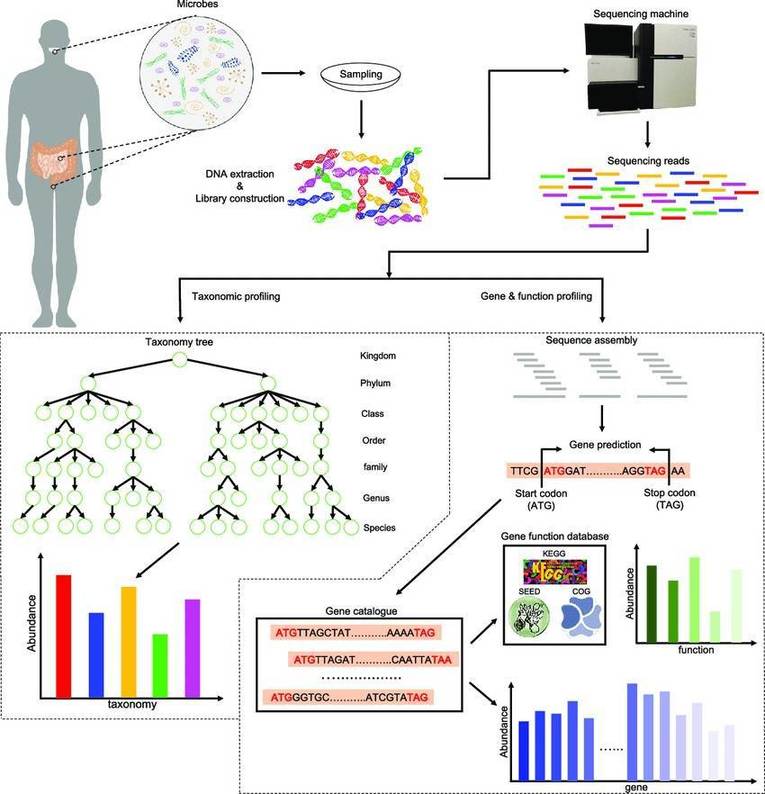
Shotgun metagenomic sequencing entails breaking ('fragmenting') DNA into many tiny chunks at random, similar to how a shotgun would tear something up into many pieces. The DNA sequences of these fragmented pieces of DNA are then stitched back together using bioinformatics to recognize the species and genes existing in the specimen. Shotgun metagenomic sequencing, unlike 16S rRNA sequencing, can read all genomic DNA in a specimen rather than just one particular area. Shotgun sequencing can simultaneously identify and profile bacteria, fungi, viruses, and a variety of other microorganisms, which is useful for microbiome research. As genomes are sequenced, microbial genes observed in the specimen (the metagenome) can be identified and profiled, providing additional information about the microbiome's functional potential. When compared to 16S rRNA sequencing, metagenomic sequencing requires a few extra steps.
A conventional shotgun metagenomics investigation entails a structured sequence of five fundamental stages: (i) acquisition, treating, and sequencing of samples; (ii) preliminary handling of sequencing data; (iii) inclusive sequence assessment to depict the taxonomic, functional, and genomic attributes of the microbiome; (iv) statistical and biological assessments for further analysis, followed by (v) validation. Initially, specimens are gathered from diverse environments or biological reservoirs (e.g., soil, water, the human gut) and processed meticulously to conserve the fidelity of microbial DNA. Subsequently, high-throughput sequencing techniques, notably the Illumina platform, are employed to generate an extensive array of concise reads.
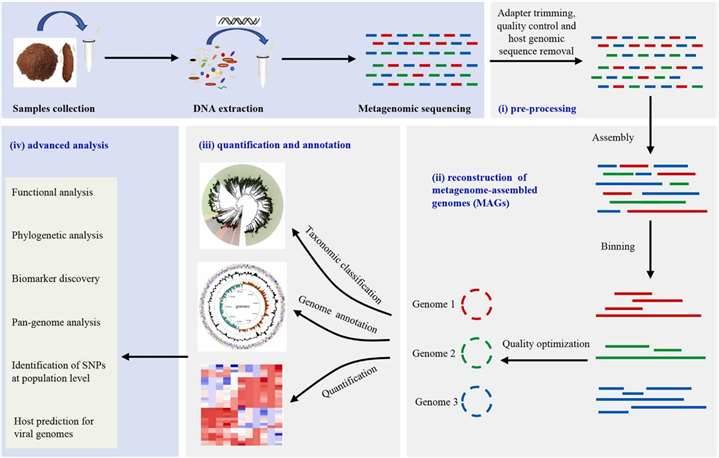 Figure 2. Typical workflow for reconstructing and analyzing metagenome-assembled genomes (MAGs) from metagenomic sequencing data. (Zhou et al., 2022)
Figure 2. Typical workflow for reconstructing and analyzing metagenome-assembled genomes (MAGs) from metagenomic sequencing data. (Zhou et al., 2022)
To evaluate the outcomes of shotgun sequencing reads, more complicated bioinformatics techniques are utilized. Quality filtering steps are also included in shotgun metagenomics bioinformatics pipelines, after which the cleaned sequencing data can be compiled to generate partial or full microbial genomes or aligned to databases of microbial marker genes. Many of these pipelines now include online tutorials and user interfaces to help people who aren't experts in bioinformatics with their analyses. The findings include information on the relative abundances of bacteria, fungi, viruses, and other microbes in the specimen, as well as curated lists of microbial genes.
Applications of Shotgun Metagenomic Sequencing
Shotgun metagenomic sequencing is a highly potent and extensively utilized technique for scrutinizing the entire genetic makeup of intricate environmental specimens. Diverging from the conventional 16S rRNA sequencing, this approach offers a more holistic and profound understanding of the composition, activities, and changes within the microbial population.
Analysis of Microbial Diversity and Community Structure
Shotgun metagenomic sequencing is an advanced technique that enables researchers to perform thorough analyses of all microorganisms present in environmental samples, surpassing the limitations inherent in targeting specific taxonomic groups. Through this approach, researchers can discern and categorize a diverse array of microbial species, gaining profound insights into their relative abundance and genetic diversity within distinct ecological niches. The seminal work of The Human Microbiome Project Consortium (2012) exemplifies the potency of shotgun metagenomic sequencing in unraveling the intricacies of microbial populations inhabiting various regions of the human body, such as the gastrointestinal tract, oral cavity, and integumentary system. By leveraging this method, the study unearthed the vast diversity and functional capabilities that underlie the human microbiome, thereby underscoring its intricate nature and the variances observed among individuals and anatomical locales. Furthermore, the research conducted by Gilbert et al. (2012) utilizing shotgun metagenomic sequencing elucidated the temporal dynamics of marine microbial communities, revealing pronounced temporal fluctuations in both community composition and metabolic potential. Such investigations underscore the versatility and power of shotgun metagenomic sequencing in unraveling the multifaceted dynamics of microbial ecosystems.
Gene Function Prediction and Metabolic Pathway Analysis
Shotgun metagenomic sequencing stands as a pivotal methodology enabling not only the identification of microbial taxa within a community but also the prediction of gene functions harbored by these microorganisms. Through the integration of genome annotation and metabolic pathway analysis, researchers can glean insights into the ecological niches and metabolic potentials of microbes inhabiting diverse ecosystems. A seminal exemplar of this technique's utility is showcased by Qin et al. (2010), who leveraged shotgun metagenomic sequencing to construct an exhaustive gene repository of the human gut microbiota. Their meticulous analysis facilitated the prediction of diverse microbial gene functions and exploration of associated metabolic pathways, illuminating the pivotal role of gut microbiota in human health and disease.
Expanding upon this foundational work, Li et al. (2014) delved deeper into the intricacies of the human gut microbiome by assembling an integrated compendium of reference genes, elucidating their functional attributes, and delineating their metabolic networks. This holistic approach not only broadens our comprehension of the microbial consortia inhabiting the human gut but also underscores the intricate interplay between microbial genetic repertoires and host physiological processes. Such endeavors underscore the indispensability of shotgun metagenomic sequencing in unraveling the enigmatic complexities of microbial communities and their profound impact on human health.
Environmental Monitoring and Ecological Research
Shotgun metagenomic sequencing serves as a cornerstone technique in environmental surveillance and ecological investigations, affording insights into the fluctuations of microbial community structures across diverse habitats. This methodology proves invaluable for discerning the ramifications of anthropogenic activities on ecosystems. Mackelprang et al. (2011) applied shotgun metagenomic sequencing to elucidate the dynamic adaptations within permafrost microbial consortia. Their study delineated an expeditious microbial response to thawing, characterized by substantial alterations in metabolic pathways, thereby underscoring its relevance to climate change studies. Likewise, Fierer et al. (2012) harnessed shotgun metagenomic sequencing to probe soil microbial populations across varied biomes, revealing distinctive functional profiles and community compositions dictated by environmental drivers.
Clinical and Public Health Research
Shotgun metagenomic sequencing is indeed emerging as a promising technique in both clinical and public health domains, offering a robust method to investigate pathogen infections, monitor the dissemination of antibiotic resistance genes, and unravel changes in microbial communities linked to various diseases. In a seminal study, Loman et al. (2013) conducted a meticulous evaluation of multiple high-throughput sequencing platforms to identify pathogens and antibiotic resistance genes present in clinical specimens. Their findings underscored the practical efficacy of shotgun metagenomic sequencing in clinical diagnostics, presenting a comprehensive approach for pathogen detection. Similarly, Schlaberg et al. (2017) further affirmed the versatility of metagenomic sequencing in detecting a diverse spectrum of pathogens in clinical samples, thereby emphasizing its crucial role in the diagnosis of infectious diseases.
Agricultural and Industrial Microbial Research
In agriculture and industry, shotgun metagenomic sequencing is employed to study soil microbes, microbial communities in fermentation processes, and more, aiming to optimize agricultural production and industrial microbial utilization. Leff et al. (2015) used shotgun metagenomic sequencing to analyze the response of soil microbial communities to increased nutrient inputs across global grasslands. The research highlighted the impact of agricultural fertilization on soil microbial communities, providing insights into sustainable agricultural practices.
Song et al. utilized next-generation sequencing to perform whole metagenome sequencing on fluids from production and injection wells, comparing them to comprehend the underground microbial community and its functions in a water-flooded reservoir. Their findings revealed that the porous environment and long-term displacement are crucial factors influencing the microbial community in reservoirs. The integration of whole metagenome analysis with the concept of porous flow holds significant implications for field studies on the geological microbial dynamics of water-flooded reservoirs.
Differences between 16s rRNA Sequencing and Shotgun Metagenomic Sequencing
16S rRNA Sequencing:
- Objective: The primary aim of 16S rRNA sequencing is the analysis of microbial community structure. The 16S rRNA gene serves as a conserved genetic marker in both bacteria and archaea, exhibiting variations between species while maintaining high conservation within species. Consequently, sequencing the 16S rRNA gene facilitates the differentiation of various microbial taxonomic units, such as genera and species.
- Method: Typically, DNA extraction from environmental samples precedes PCR amplification of specific regions of the 16S rRNA gene. Subsequently, these PCR products undergo sequencing, with the resultant sequences compared and classified using specialized 16S rRNA databases like Greengenes or SILVA.
- Advantages: The technique offers a relatively low-cost solution, rendering it suitable for high-throughput analysis of large sample sets. Additionally, it furnishes relative abundance information pertaining to microbial community structure.
- Limitations: Despite its utility in providing taxonomic information, 16S rRNA sequencing lacks functional insights. Moreover, it is susceptible to PCR biases, potentially compromising result accuracy. Furthermore, its inability to identify to the species level limits its taxonomic classification to a coarse level.
Shotgun Metagenomic Sequencing:
- Objective: Shotgun metagenomic sequencing endeavors to sequence the entire genomic content of microbial communities, thereby furnishing comprehensive genetic information about all organisms therein.
- Method: This methodology involves direct extraction of DNA from samples, followed by random fragmentation. These fragments subsequently undergo sequencing, with bioinformatics tools employed to process the resulting data, identifying and annotating genes from diverse microorganisms.
- Advantages: The approach provides exhaustive genetic information encompassing functional and metabolic potential. Moreover, it is adept at identifying and characterizing all microorganisms present, spanning bacteria, archaea, fungi, and viruses.
- Limitations: Despite its efficacy, shotgun metagenomic sequencing commands a higher cost, particularly for large-scale sample sets. Furthermore, its data analysis and interpretation necessitate advanced bioinformatics skills. Additionally, the comprehensive nature of sequencing results in substantial data volumes, mandating significant computational resources for processing and analysis.
The primary distinctions between 16S rRNA Sequencing and Shotgun Metagenomic Sequencing lie in the following aspects:
Difference in sequencing throughput:
16S rRNA Sequencing and Shotgun Metagenomic Sequencing exhibit a sequencing throughput difference of three orders of magnitude. For example, sequencing the 16S V3-V4 region of bacteria using Miseq 2300bp typically yields around 30,000 reads, amounting to approximately 18 million data points. Conversely, shotgun metagenomic sequencing on platforms like Nova seq with 2150bp reads can generate 10 gigabytes of data, equivalent to 10,000 million bases, covering bacteria, fungi, archaea, and more.
Divergence in sequenced regions:
Shotgun metagenomic sequencing enables sequencing within a 5 million base pair (5M) region of bacteria, holding vast potential for detailed analysis. Comparatively, full-length sequencing of bacteria's 16S rRNA gene typically spans around 1.5 thousand base pairs (1.5K or 1500bp). Opting for sequencing the V3-V4 region of bacterial 16S narrows the length of sequenced data to approximately 470bp.
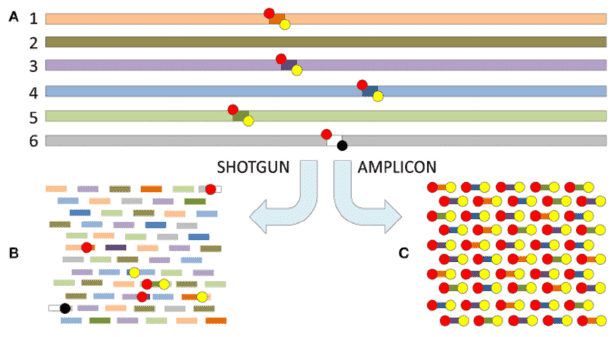
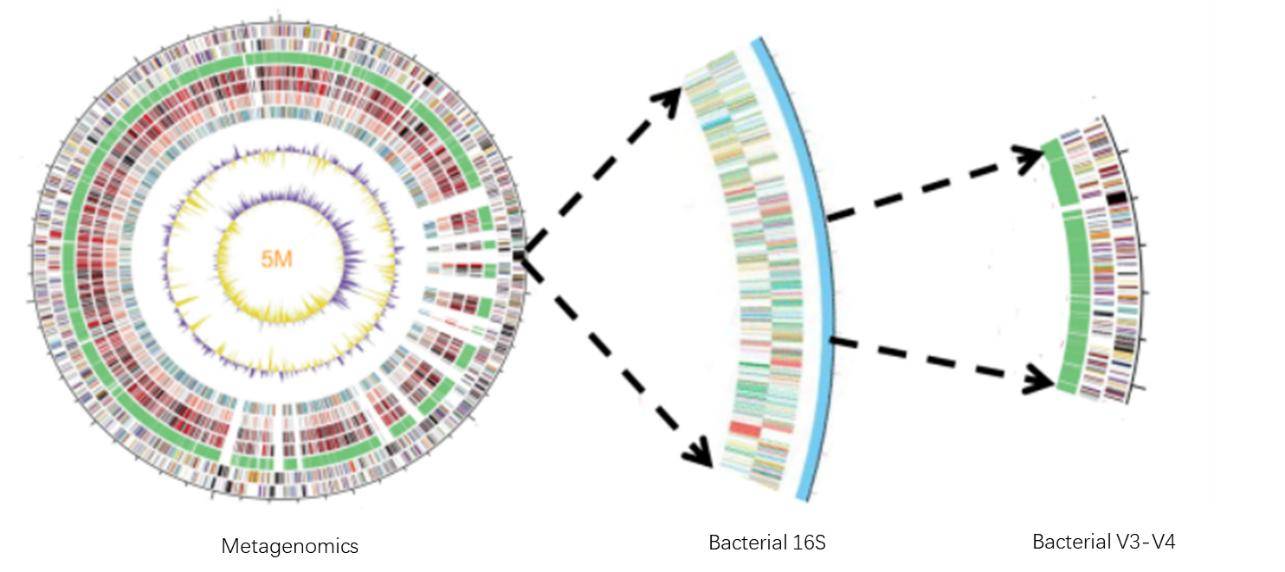 Figure 3. Comparison of Target Regions in Metagenomics and 16S Sequencing.
Figure 3. Comparison of Target Regions in Metagenomics and 16S Sequencing.
Content analysis differences:
16S rRNA sequencing primarily investigates the species composition, evolutionary relationships between species, and diversity within a community. In contrast, shotgun metagenomic sequencing goes beyond 16S analysis to delve into gene and functional aspects.
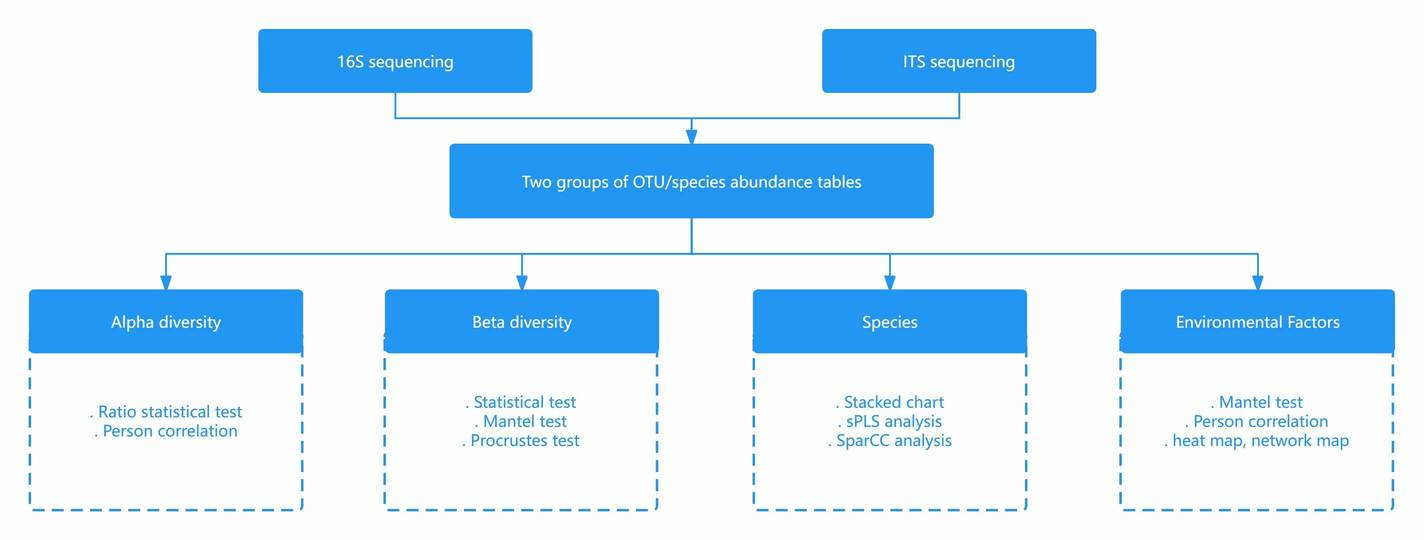
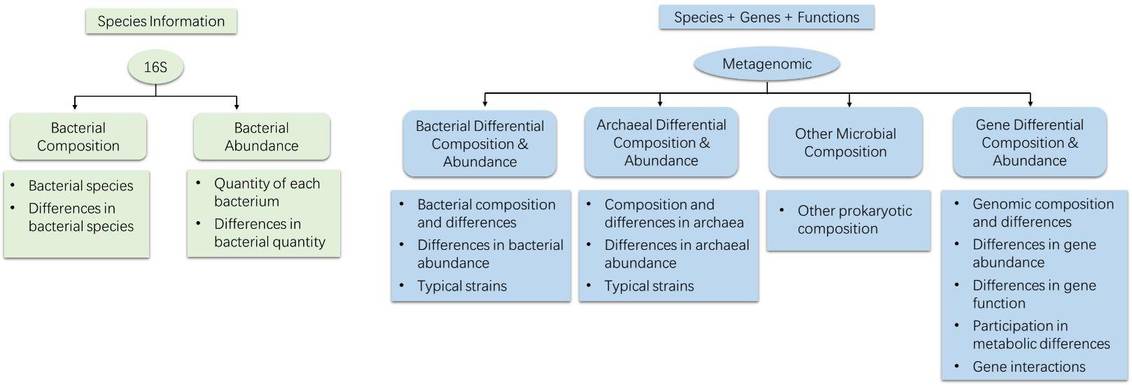 Figure 4. Comparison of Analytical Content in Metagenomics and 16S Sequencing.
Figure 4. Comparison of Analytical Content in Metagenomics and 16S Sequencing.
For example, Chen et al. (2019) examined the impact of a certain drug on the intestinal microbiota of rats using 16S sequencing. They conducted quality assessment of the sequencing data, alpha diversity analysis, species annotation analysis, beta diversity analysis, and microbial functional prediction.
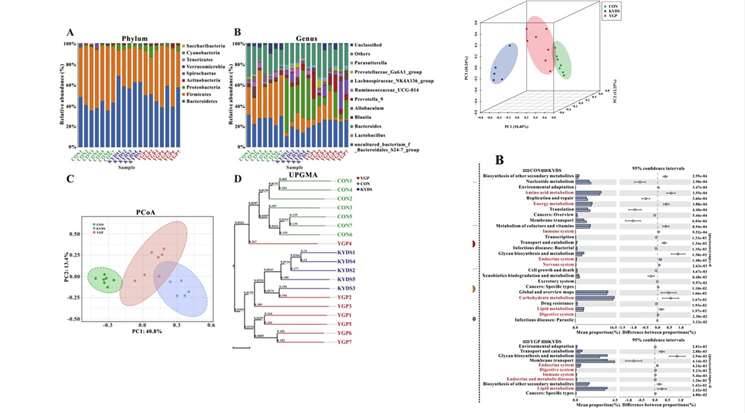 Figure 5. Case Studies of Analytical Results from 16S Sequencing. (Chen et al., 2019)
Figure 5. Case Studies of Analytical Results from 16S Sequencing. (Chen et al., 2019)
On the other hand, Tong et al. (2022) studied the microbial communities associated with a water buffalo's digestive system using shotgun metagenomic sequencing. They obtained metagenome-assembled genomes (MAGs) at the strain level for 4960 strains and at the species level for 3255 strains. Through LEfSe analysis, they identified 359 differentially abundant taxa at the genus level. Additionally, by annotating the protein-coding genes of MAGs and comparing them with the CAZy and eggNOG databases, they assessed the microbial ecological functions of MAGs in the digestive tract.
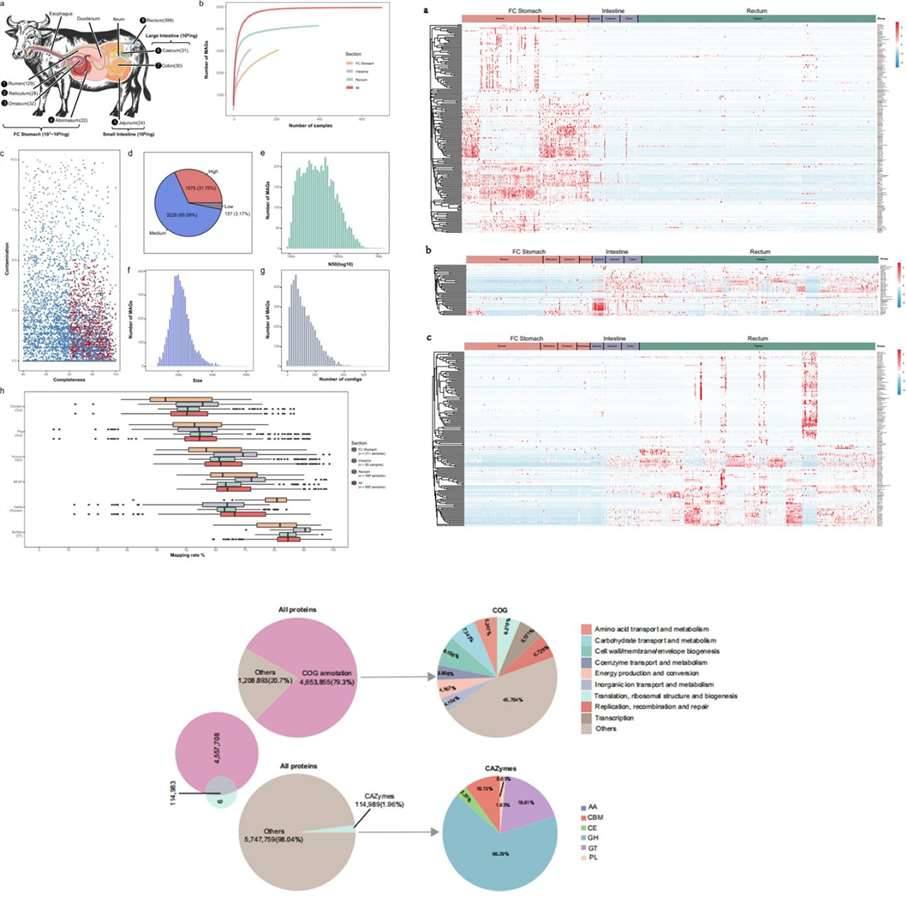 Figure 6. Case Studies of Analytical Results from Metagenomic Sequencing. (Tong et al., 2022)
Figure 6. Case Studies of Analytical Results from Metagenomic Sequencing. (Tong et al., 2022)
Table 1. 16s rRNA Sequencing VS Shotgun Metagenomic Sequencing
| Factors | 16s rRNA Sequencing | Shotgun Metagenomic Sequencing |
|---|---|---|
| Functional profiling (profile microbial genes) | No (although 'predicted' functional profiling is an option). | Yes, indeed (but it only reveals information on functional potential) |
| Taxonomic resolution: Genus, species, strain | Bacterial genus (or species), depending on location (s) centered | Species of bacteria (sometimes strains and single nucleotide variants, if sequencing is deep enough) |
| Taxonomic coverage | Bacteria and archaea are two types of bacteria. | Viruses are included in all taxa. |
| Bioinformatics requirements | Expertise ranging from beginner to intermediate | Intermediate to advanced level of knowledge |
| Databases | Established and carefully curated | Comparatively new, still improving |
| Sensitivity to host DNA contamination | Low (but PCR success according on the absence of inhibitors and the presence of a detectable microbiome) | High, but differs depending on the sample kind (but this can be mitigated by calibrating the sequencing depth) |
| Bias | High-to-medium (acquired taxonomic composition is dependent on selected primers and targeted variable area) | Lower (while metagenomics is "untargeted," experimental and analytical biases can be proposed at any point) |
How to Choose Between 16S rRNA Sequencing and Shotgun Metagenomic Sequencing
When it comes to microbial detection, whether to choose 16S rRNA sequencing or shotgun metagenomic sequencing poses a crucial decision.
- If the aim is to quickly understand the overall situation of the microbial community under study, 16S sequencing is preferable.
- For those seeking in-depth insights into detailed microbial strain-level information and disease risk-related data, even if time and cost are not a constraint, opting for shotgun metagenomic sequencing is recommended.
- Analyzing the compositional differences and characteristics of microbial community structures calls for 16S sequencing.
- When investigating microbial community structures, species classification, gene functions, and metabolic networks, shotgun metagenomic sequencing is the method of choice.
- Employing a research strategy that combines 16S and shotgun metagenomic sequencing involves conducting 16S sequencing on a large sample size and subsequently selecting representative samples for shotgun metagenomic sequencing based on differential species.
While 16S sequencing enables rapid and accurate identification of microbial species and quantities, typically at the genus level, fine resolution to the species or subspecies level may be challenging. Shotgun metagenomic sequencing provides comprehensive genomic information of microorganisms, allowing identification at the species or subspecies level, albeit at a higher sequencing cost compared to 16S sequencing. Therefore, the selection of a microbial detection method should factor in research objectives and budgetary considerations.
References
- Durazzi F, Sala C, Castellani G, et al. Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota. Scientific reports. 2021, 4;11(1).
- Mori H, Maruyama T, Yano M, et al. VITCOMIC2: visualization tool for the phylogenetic composition of microbial communities based on 16S rRNA gene amplicons and metagenomic shotgun sequencing. BMC systems biology. 2018 ,12(2).
- Patel SR, Ingram C, Scovell JM, et al. The microbiome and urolithiasis: current advancements and future challenges. Current Urology Reports. 2022, 23(3):47-56.
- Chiu CY, Miller SA. Clinical metagenomics. Nature Reviews Genetics. 2019, 20(6):341-355.
- Yan Z, Yang F, Cao J, et al. Alterations of gut microbiota and metabolome with Parkinson's disease. Microbial Pathogenesis. 2021, 160:105187.
- Delgado-Baquerizo M, Eldridge D J, Liu Y R, et al. Global homogenization of the structure and function in the soil microbiome of urban greenspaces. Science Advances, 2021, 7(28): eabg5809.
- Dargiri S A, Movahedi A. Diversity and biogeography of soil bacterial communities//Climate Change and Microbiome Dynamics: Carbon Cycle Feedbacks. Cham: Springer International Publishing, 2023: 1-13.
- Fan H, He P, Xu S, et al. Banana disease-suppressive soil drives Bacillus assembled to defense Fusarium wilt of banana. Frontiers in Microbiology, 2023, 14: 1211301.
- Bulgarelli D, Rott M, Schlaeppi K, et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 2012, 488(7409):91-95.
- Cao S, Du R, Li B, et al. High-throughput profiling of microbial community structures in an ANAMMOX-UASB reactor treating high-strength wastewater. Applied Microbiology and Biotechnology, 2016, 100: 6457-6467.
- Kochetkova T V, Grabarnik I P, Klyukina A A, et al. Microbial communities of artisanal fermented milk products from Russia. Microorganisms, 2022, 10(11): 2140.
- Kormas K A, Meziti A, Mente E, et al. Dietary differences are reflected on the gut prokaryotic community structure of wild and commercially reared sea bream (Sparus aurata). Microbiologyopen, 2014, 3(5): 718-728.
- Suchodolski JS. Intestinal microbiota of dogs and cats: a bigger world than we thought. Veterinary Clinics of North America: Small Animal Practice. 2011, 41(2):261-272.
- Emmons A L, Mundorff A Z, Keenan S W, et al. Characterizing the postmortem human bone microbiome from surface-decomposed remains. PloS one, 2020, 15(7): e0218636.
- Lax S, Smith DP, Hampton-Marcell J, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science. 2015, 345(6200):1048-1052.
- Quince C, Walker AW, Simpson JT, et al. Shotgun metagenomics, from sampling to analysis. Nature biotechnology. 2017, 35(9):833-44.
- Zhou Y, Liu M, Yang J. Recovering metagenome-assembled genomes from shotgun metagenomic sequencing data: methods, applications, challenges, and opportunities. Microbiological Research, 2022, 260: 127023.
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature, 2012, 486, 207-214.
- Gilbert J A, Steele J A, Caporaso J G, et al. Defining seasonal marine microbial community dynamics. The ISME journal, 2012, 6(2): 298-308.
- Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. nature, 2010, 464(7285): 59-65.
- Li J, Jia H, Cai X, et al. An integrated catalog of reference genes in the human gut microbiome. Nature biotechnology, 2014, 32(8): 834-841.
- Fierer N, Leff J W, Adams B J, et al. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proceedings of the National Academy of Sciences, 2012, 109(52): 21390-21395.
- Mackelprang R, Waldrop M P, DeAngelis K M, et al. Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature, 2011, 480(7377): 368-371.
- Loman N J, Misra R V, Dallman T J, et al. Performance comparison of benchtop high-throughput sequencing platforms. Nature biotechnology, 2012, 30(5): 434-439.
- Schlaberg R, Chiu C Y, Miller S, et al. Validation of metagenomic next-generation sequencing tests for universal pathogen detection. Archives of Pathology and Laboratory Medicine, 2017, 141(6): 776-786.
- Leff J W, Jones S E, Prober S M, et al. Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe. Proceedings of the National Academy of Sciences, 2015, 112(35): 10967-10972.
- Song Z, Chen S, Zhao F, et al. Whole metagenome of injected and produced fluids reveal the heterogenetic characteristics of the microbial community in a water-flooded oil reservoir. Journal of Petroleum Science and Engineering, 2019, 176: 1198-1207.
- Chen R, Wang J, Zhan R, et al. Fecal metabonomics combined with 16S rRNA gene sequencing to analyze the changes of gut microbiota in rats with kidney-yang deficiency syndrome and the intervention effect of You-gui pill. Journal of ethnopharmacology, 2019, 244: 112139.
- Tong F, Wang T, Gao N L, et al. The microbiome of the buffalo digestive tract. Nature Communications, 2022, 13(1): 823.