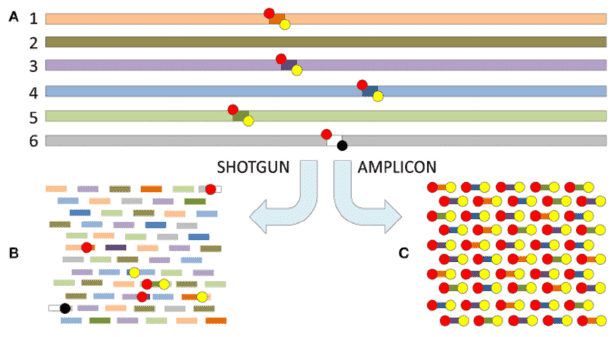
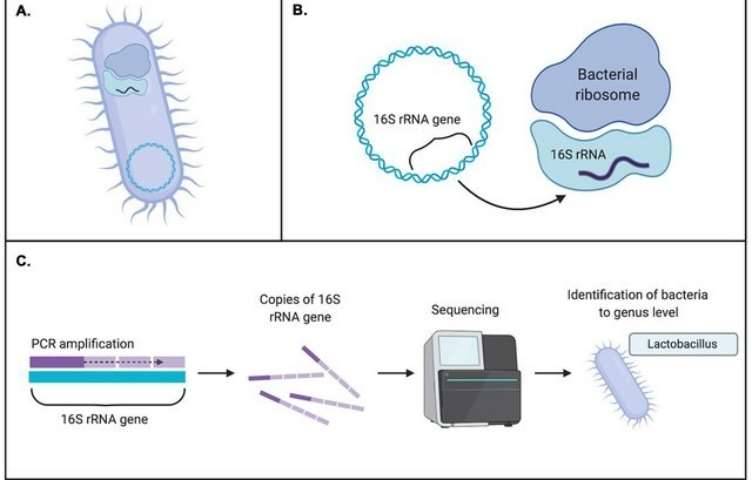
Microorganisms represent the smallest entities within ecosystems, yet their significance in these systems is paramount. Their influence extends to the biodiversity of soil, water, and air, with consequential effects on human health and disease. Consequently, the detection and study of microorganisms emerge as vital pursuits in scientific research.
Traditional methods in microbial research, such as pure cultivation and physicochemical analysis, face limitations when dealing with the majority of microorganisms in natural environments—many of which are non-culturable and challenging to identify. Advancements in technologies like high-throughput sequencing and mass spectrometry have led to the development and application of diverse microbial detection methods. Among these, 16S rRNA gene sequencing and metagenomic sequencing stand out as commonly employed techniques. Both approaches contribute significantly to the exploration of microbial diversity and functionality within ecosystems.
Introduction to 16S rRNA Sequencing
The 16S rRNA gene is universally present in all bacteria and archaea. It spans approximately 1500bp, comprising 9 highly variable regions and 10 conserved regions. 16S amplicon sequencing methods exploit the 16S rRNA gene by amplifying through PCR to obtain either partial or full length sequences of the gene. These sequence data provide the means to identify the species and abundance of microbes, as well as investigate the structure and diversity of microbial communities.
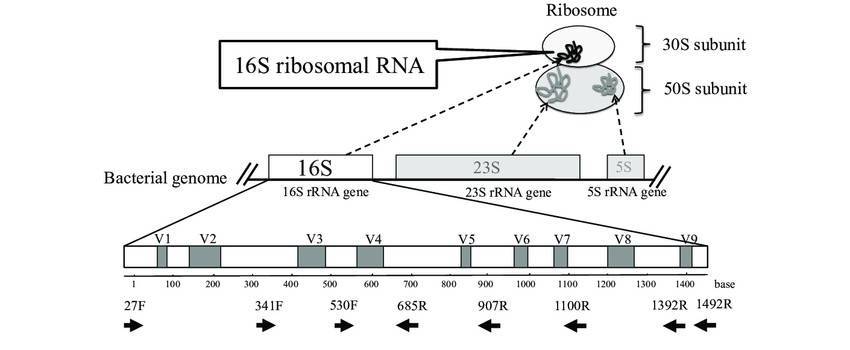 Fig. 1. The schema of ribosome complex and 16S rRNA gene. (Kazumasa Fukuda et al,.2016)
Fig. 1. The schema of ribosome complex and 16S rRNA gene. (Kazumasa Fukuda et al,.2016)
Sequencing Platform and Sequencing Strategy
Generally speaking, the lengthier the sequencing fragment, the more accurate the species identification. However, choices of sequencing platforms and areas to amplify are often determined by the objective of the analysis, which is susceptible to constraints like sequencing platforms and costs involved.
Employing traditional sanger sequencing methods offer benefits such as high accuracy, relatively low cost, and long sequencing reads, nearly covering the full length of the 16S rRNA gene. The limitation lies in its inability to sequence and analyze complex or mixed samples directly, therefore leading to lower sequencing throughput. Leveraging high-throughput sequencing technology, or next-generation sequencing (NGS), which can directly construct libraries and sequence complex samples, has become an increasingly popular approach in the current research surrounding 16S amplicon sequencing. Drawbacks include shorter read lengths, typically confining amplification to specified regions like V1-V3, V3-V4, or V4-V5, followed by sequencing analysis conducted in combination with PE250 or PE300.
The long read sequencing technology stands out due to its ultralong read lengths and high-throughput capabilities, facilitating the facile acquisition of the full length of 16S rRNA genes. However, its higher error rate and cost serve as demerits, which have hindered its wider application. It is expected that as this technology continues to evolve and become more refined, long read sequencing may emerge to be the foremost method employed in microbial research.
Amplification Region Chosen for 16S Amplicon Sequencing
Among the varying methods currently used for 16S amplicon sequencing, next-generation high-throughput sequencing is the most commonly employed. However, the need for choosing the amplification region arises due to the constraint of sequencing read length. The Earth Microbiome Project (EMP) recommends the standard primers for amplifying the V4-V5 regions (515F/806R primer pair) and the V4-V6 regions (515F/926R primer pair) (Table 1). Some researchers have evaluated commonly used primers in 16S amplicon sequencing and found that the V4–V6 regions of the 16S rRNA gene provides good specificity and complete database information. The EMP recommended primers, credited for their high coverage and specificity, have emerged as the best choice for annotating bacterial diversity analysis.
For further insights, refer to the emcompassing review titled "16S/18S/ITS Amplicon Sequencing Overview".
Table 1 Basic information of 16S rRNA gene universal primers
| No. | Primers name | Sequences (5'→3') | Amplified region | Amplified fragment length (bp) |
|---|---|---|---|---|
| 2 3 4 5 6 7 8 |
27F 342R 355F 519R 355F 806R 515F 806R 515F 926R 515F 1064R 1114F 1392R 27F 1492R |
AGRGTTYGATYMTGGCTCAG CTGCTGCSYCCCGTAG ACWCCTACGGGWGGCWGC GWATTACCGCGGCKGCTG ACWCCTACGGGWGGCWGC GGACTACNVGGGTWTCTAAT GTGYCAGCMGCCGCGGTAA GGACTACNVGGGTWTCTAAT GTGYCAGCMGCCGCGGTAA CCGYCAATTYMTTTRAGTTT GTGYCAGCMGCCGCGGTAA AYCTCACGRCACGAGCTGAC GCAACGAGCGCAACCC ACGGGCGGTGTGTRC AGRGTTYGATYMTGGCTCAG TACGGYTACCTTGTTACGACTT |
V1-V2 V3 V3-V5 V4-V5 V4-V6 V4-V7 V9 V1-V10 |
315 164 451 291 411 550 278 1465 |
Comparison of 16S amplicon sequencing and metagenomic sequencing
As a definitive divergence from 16S amplicon sequencing, metagenomic sequencing involves directly sequencing all genes present in microbial DNA. This approach allows researchers to obtain the whole genome sequence of microorganisms. By analyzing these sequences, one can study the genomic structures and functions of microorganisms. Metagenomic sequencing can be utilized in research seeking to explore microbial metabolic pathways, antibiotic resistance, and toxicity along with other functional aspects. Both 16S amplicon sequencing and metagenomic sequencing present their unique advantages and limitations. 16S amplicon sequencing assists in quickly and accurately identifying the types and quantities of microorganisms, but it typically only allows for identification up to the genus level, and does not permit accurate specification to species or subspecies levels. On the other hand, metagenomic sequencing yields the entire genomic information of microorganisms, enabling identification down to species or subspecies levels. However, in comparison to 16S sequencing, it is associated with higher sequencing costs. Therefore, in the selection of microbial detection methods, one must weigh the research objectives and budget considerations to choose the most appropriate method.
For more information of comparative analysis between metagenomic sequencing and amplicon sequencing, one may refer to the article titled "Amplicon-Based Next-Generation Sequencing vs. Metagenomic Shotgun Sequencing."
Introduction to Metagenomic Sequencing
Let's focus on metagenomics. Metagenomic sequencing entails high-throughput sequencing of all microbial DNA present within a sample. Subsequent sequence processing and analysis can identify the species composition of microbial communities, trace their evolutionary relationships, and even explore functional activity of microbial cohorts, investigating molecular mechanisms behind pathogenicity or antibiotic resistance, and unearthing molecular pathways of research value. The workflow for metagenomic sequencing and analysis (Fig. 2) consists of sample collection, DNA extraction, library construction and sequencing, sequence assembly, binning, abundance and diversity analysis, and custom-tailored advanced analyses.
For further reading, consider the reference article titled "Overview of Metagenomics Sequencing".
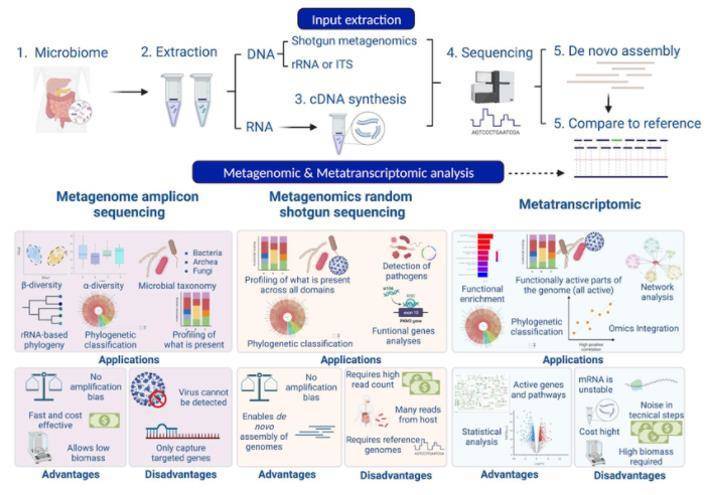 Fig. 2. Workflow of a metagenomic analysis: laboratory and bioinformatic analysis (Laura C. Terrón-Camero et al,. 2022)
Fig. 2. Workflow of a metagenomic analysis: laboratory and bioinformatic analysis (Laura C. Terrón-Camero et al,. 2022)
Application of 16S rRNA and Metagenomic Sequencing
The 16S amplicon sequencing and metagenomic sequencing methodologies both have extensive applications. For instance, 16S amplicon sequencing can be employed to investigate the diversity and composition of microbial communities in varied environments such as soil, water bodies, and animal intestines. Metagenomic sequencing, on the other hand, can be utilized to explore functional aspects of the microbiome like metabolic pathways, antibiotic resistance, and toxicities diverse scenarios like studying the impact of gut microbiota on human health, or the degradation capabilities of environmental microorganisms.
In addition to 16S amplicon and metagenomic sequencing, there are other microbial detection strategies, such as fluorescence in situ hybridization (FISH) and quantitative PCR (qPCR). Each method has its own strengths and weaknesses and should be chosen based on the objectives of the research and available budget.
In summary, microbial detection and research are integral for both ecosystems and human health. 16S amplicon sequencing and metagenomic sequencing are two commonly employed microbial detection methods, which are capable of exploring microbial diversity and function. When selecting the detection method, one should choose appropriately based on their research objectives and budget constraints.
Reference
- Yunyan Zhou, Min Liu, Jiawen Yang, Recovering metagenome-assembled genomes from shotgun metagenomic sequencing data: Methods, applications, challenges, and opportunities, Microbiological Research, Volume 260,2022,127023,ISSN 0944-5013.