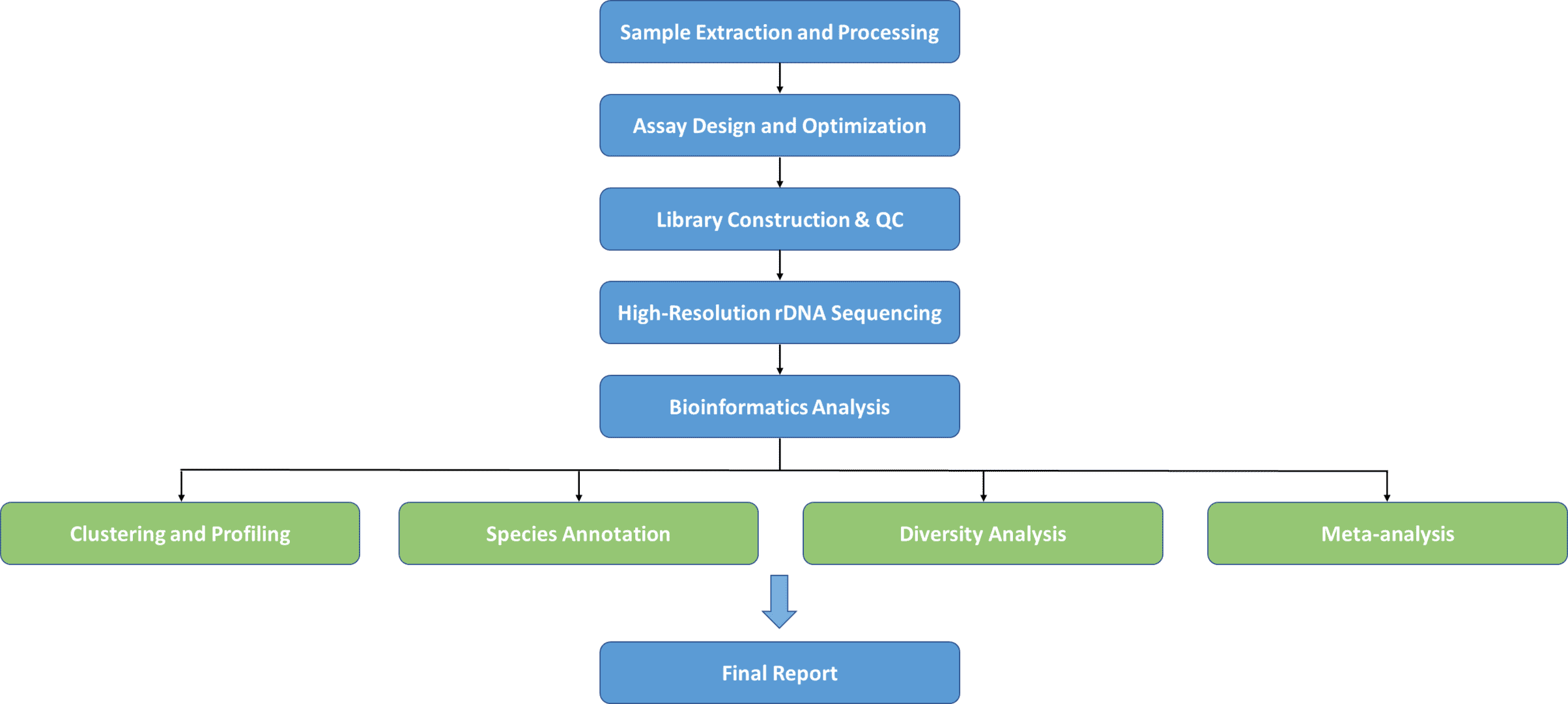
Between distinct species of microorganisms, containing nine hypervariable areas varying from about 30 to 100 base pairs long, the rRNA gene is highly preserved, varying significantly between bacteria. Highly conserved areas enable scientists to construct primer pairs that will augment the hypervariable area of their preference accurately and reliably in order to identify or characterize various bacterial communities. rRNA gene sequencing is the best instrument for studying bacterial and fungal taxonomy and molecular phylogeny, depending on the advancement of sequencing innovation. CD Genomics offers a full-length rRNA sequencing service to help assist your studies by taking advantage of PacBio SMRT long-reads sequencing methods.
The method provides numerous huge benefits, such as high throughput bioinformatic pipelines and reference databases founded, and low cost per specimen. The use of this strategy has indicated the microbiota's remarkably varied character and substantially developed our knowledge of its role in various body areas in human health and disease.