Next-Generation Sequencing of Viral Genomes: Introductions, Technologies, and Applications
Inquiry >Viruses pose great threats to public health worldwide. Identifying viruses and conducting epidemiological investigations rapidly and accurately is, therefore, of great significance for epidemiologists and healthcare providers. In order to address the needs of disease control and precaution, especially during viral outbreaks, next-generation sequencing (NGS) has become a useful tool for an efficient and effective understanding of virus pathogenicity and epidemiology by providing the sequences of pathogen genomes, which can further contribute to the development of novel therapies and vaccines.
The recent advances of NGS have enormously changed the field of virus discovery. NGS can be a versatile tool. Several approaches are commonly used in virology studies, including amplicon-based NGS, hybrid capture-based sequencing, metagenomic sequencing, and whole-genome sequencing (WGS). Depending on the requirements of the specific project, different approaches should be selected.
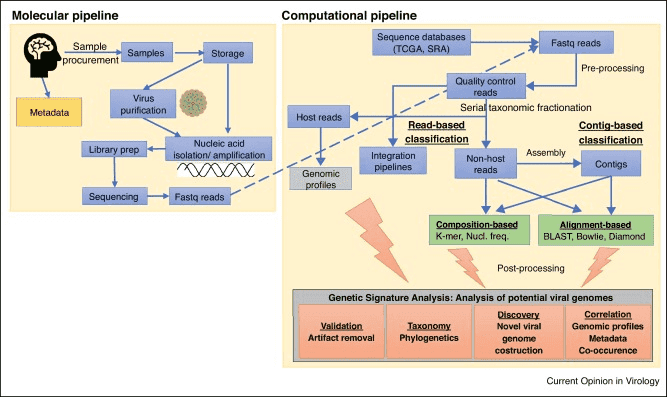 Figure 1. Detecting viral sequences in NGS data (Cantalupo 2019)
Figure 1. Detecting viral sequences in NGS data (Cantalupo 2019)
Amplicon-Based NGS
Amplicon sequencing is an efficient and cost-effective method for accurate measurement of virus diversity. It uses primers that target a specific gene region of interest, followed by NGS. The targeted region typically contains a highly variable region, allowing for detailed identification and the determination of genetic variation in specific genomic regions and the phylogenies of a sample. However, primers do not have the same affinity for all possible sequences, so bias can occur during amplification. Optimization of primer selection is helpful to reduce bias. Prior knowledge on viral genome is required to evaluate the taxonomic resolution and coverage of the targets. Thus, amplicon sequencing is ideal for a small number of well-defined regions.
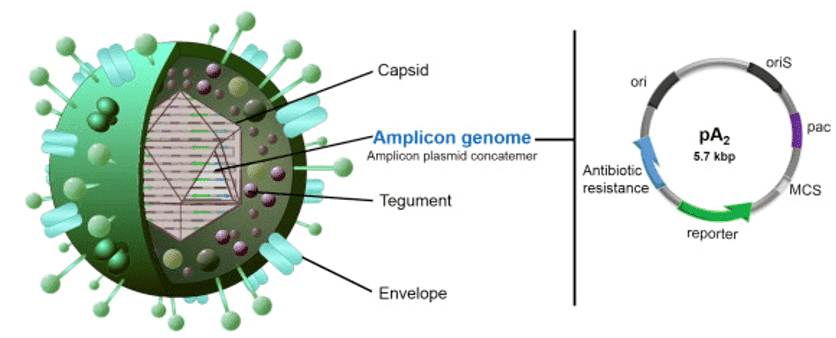 Figure 2. Viral amplicon vector and amplicon plasmid structure. (Baez 2018)
Figure 2. Viral amplicon vector and amplicon plasmid structure. (Baez 2018)
Shotgun Sequencing
Compared to amplicon-based sequencing, shotgun sequencing uses random shearing without selective enrichment of a specific population, allowing for both single-species WGS and metagenomic analysis. This method captures all potential pathogen genes present in the sample, including viral, bacterial, and eukaryotic genes, thus, yielding more detailed genomic information and taxonomic resolution and allowing researchers to comprehensively analyze all genes in a complex sample. With adequate sequencing depth, the assembly of entire viral genomes from short sequences can be done and the taxonomic resolution to species or strain level can be reached. DNA and RNA viruses can be detected by uniquely identifying DNA and/or RNA shotgun sequences.
Hybrid Capture-Based Sequencing
A hybridization assay is another category of enrichment assay other than PCR amplification. The hybridization-based capture is applied after nucleic acid extraction and library preparation. The protocols start with random shearing of the genes, denatured by heating. Randomly sheared overlapping fragments are captured by DNA or RNA single-stranded oligonucleotides specific to the region of interest (hybridization). The non-specific unbound molecules are washed away, and the enriched nucleic acids are sequenced by NGS. This allows for independent sequencing of a large number of unique fragments of targeted regions. Any duplicates can be easily identified and removed, leaving high-quality data for analysis.
References
- Deng X; et al. Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance. Nature Microbiology. 2020, 5(3): 443-54.
- Grubaugh ND; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biology. 2019 20(1): 8.
- Cantalupo PG; Pipas JM. Detecting viral sequences in NGS data. Current opinion in virology. 2019, 39:41-8.
- Baez MV; et al. Using Herpes Simplex Virus Type 1-Based Amplicon Vectors for Neuroscience Research and Gene Therapy of Neurologic Diseases. Molecular-Genetic and Statistical Techniques for Behavioral and Neural Research: Elsevier; 2018.