Microbiome and Cancer
Alterations in the microbiome may lead to cancer. For different tissue types, the responsibilities of each microbial species in cancer development have previously been known. In tissues directly exposed to microbes, localized inflammation has often been identified. Optionally, where the bacterial community had a centralized protective effect, certain tissues were recognized. In addition, in the lack of microbial materials, some immune-modulatory treatments and standard therapies depend on the inflammatory response that is repressed.
More lately, findings have begun that describe microbiomes that act at a distance to affect sterile tumor areas and can affect both natural autoimmunity and anticancer therapies that modulate immune functions. Reaction to CpG-oligonucleotide immunotherapy and platinum chemotherapy in sterile or antibiotic-treated mice with poor myeloid-derived tumor infiltration and low production of cytokines was affected. Cross-reactivity between microbial antigens and tumor antigens may have been influenced by systemic effects affecting the T-cell repertoire.
Microbiome Effects on Immunotherapy
The advantage of evaluating microbiomes along with immunotherapy methods has been shown by recent research in mouse models, finding that key organisms in the gut microbiome assert systemic impacts on the effectiveness of immune-modulatory drugs. Particular bacteria that impair the capacity of checkpoint inhibitor drugs to enhance immune response have been involved in two studies. In one study, the loss of a specific gut bacteria was related to a negative end result using CTLA-4 blockade treatment. The outcome was enhanced, however, with several combinatorial methods, such as bacterial gavage, the use of bacterial immunization antigens, or the adoptive transfer of antigen-specific T-cells. To recognize another microbe that influenced the effects of anti-PD-L1 treatment, an independent study utilized 16S rRNA sequencing. Equally, a combinatorial process greatly inhibits the growth of the tumor affiliated with T cell buildup in the microenvironment of the tumor. Together, these researches illustrate the significance of classifying advantageous organisms and using microbiota-manipulation combinatorial methods.
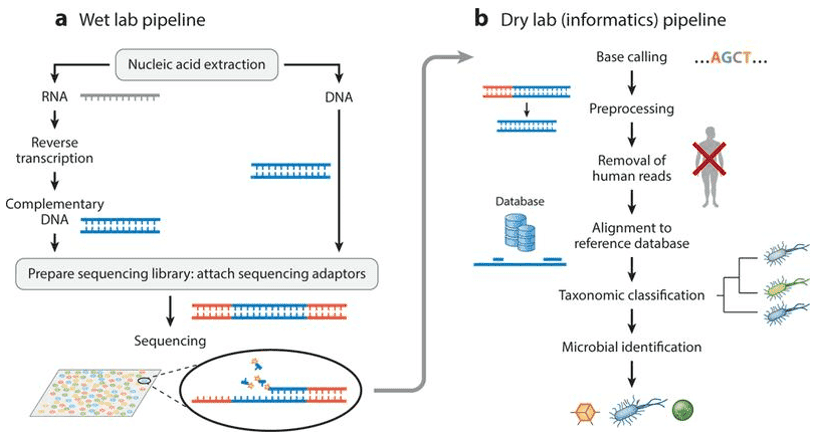
Methods for Microbiome Profiling
Sequencing 16S ribosomal RNA (rRNA) is a technique of sequencing focused on amplicons that identify a genetic marker found in all bacteria. Bacteria present within a given specimen down to the level of the genus and/or species are usually utilized to identify them. 16S rRNA gene sequencing is a well-established technique for analyzing phylogeny and taxonomy of samples from complex microbiomes or environments.
Metagenomic Shotgun Sequencing
For species recognition and functional evaluation, shotgun metagenomic sequencing is employed to sequence all the genomic information in a microbial specimen. Shotgun metagenomic sequencing, with high sequence availability, can trace rare and low-abundance microbial community members. The technique allows scientists in different environments to assess microbial diversity and classify the richness of microbes.
Metatranscriptomes encapsulate all RNAs embedded in a complex data set by a group of specimens. Metatranscriptome analysis appeals microbial samples to RNA sequencing (RNA-Seq) to evaluate which organisms are there, what they convey, and how they react to environmental changes. Microbial RNA-Seq allows non-biased strand-specific detection of common and novel transcripts, in contrast to hybridization-based techniques. In order to evaluate gene expression mutations, anticipate antibiotic resistance, comprehend host-pathogen relationships, and trace disease progression, metatranscriptome data can be employed.
References
- Candela M, Turroni S, Biagi E, et al. Inflammation and colorectal cancer, when microbiota-host mutualism breaks. World journal of gastroenterology: WJG. 2014 Jan 28;20(4).
- Schwabe RF, Jobin C. The microbiome and cancer. Nature Reviews Cancer. 2013 Nov;13(11).