A large part of bacterial, archaeal, viral, and fungal microbial taxa consists of the human microbiome. While many of these microorganisms are commensal, some are harmful to humans and many are symbiotic. Our livelihoods are strongly intertwined with the microbes we allocate our bodies with, despite whether their appearance is advantageous, insignificant, or harmful. Over the past few years, human microbiome research, defined as the research of the whole DNA component of micro-organisms that reside in our bodies, has grown quickly. Blood has always been considered free of microorganisms. But recent DNA sequencing methods have shown that 1 mL of blood contains about 1,000 bacterial cells. These bacteria are usually dormant. The profiling and characteristics of the blood microbiome by methods such as next-generation sequencing and long-read sequencing can facilitate the research on many diseases including bacterial infections and even cancer.
Next-Generation Sequencing Facilitating Blood Microbiome Research
The emergence of Next Generation Sequencing (NGS) especially the invention of whole metagenome shotgun sequencing (WMGS) as methods to analyze microbial genetic material existing in various human body parts has mainly promoted the discovery of our "microbial-selves." Researchers have sought to determine a taxonomy-based set of core human-associated microorganisms as well as to compare pathogenesis with variations, modifications, and even the dysbiosis of microbiome. Numerous large population-based researches have synthesized the human intestinal microbiome metagenomes in this aspect, as well as other specific medical body sites, such as the gut, skin, vagina, mouth, and blood.
Recent studies based on high-throughput sequencing of 16S rDNA have demonstrated the presence of multiple microbiomes in the blood of patients with sepsis and healthy individuals. Although its biological and clinical significance remains to be further explored, the discovery of the blood microbiome may be an important step toward a better understanding of the human microbial world and its relationship to health and disease. Therefore, there is an urgent need to use culture-independent next-generation sequencing technologies to determine whether patients' blood is rich in microbiota to better understand the development of bacteraemia, infectious complications, and other diseases.
A Workflow for Human Blood Microbiome Characterization
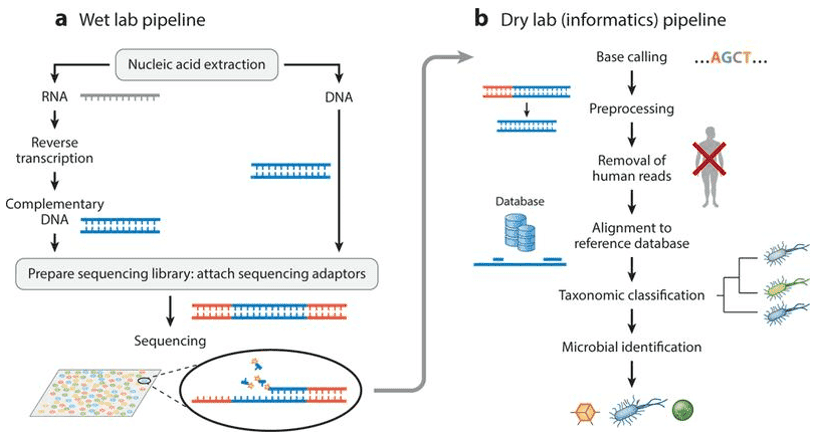
In a typical workflow for human blood microbiome characterization, by amplification and sequencing of the bacterial 16S ribosomal gene using oligonucleotide primers which aim variable region 4, the circulating microbiome is evaluated at the DNA level. Direct amplification of the V4 region is conducted alongside a negative control response that underwent the full experimental procedure. Triplicate amplification is conducted as 20 μl reactions containing; 1.0 μl plasma, 10 μl 2X PCR buffer, 0.4 μl DNA polymerase, 1.0 μl of each forward and reverse oligonucleotide primer (10 μM).
Before a further PCR, amplicons leading from triplicate reactions are merged and cleaned, and a pair of V4 oligonucleotide primers to integrate the adapters in sequencing preparation. Then the PCR cycling is conducted and the PCR products are purified at a ratio of 0.8 beads to sample (v/v), eluted to 20 μl molecular biology grade water. Employing the Illumina MiSeq system with a 250 bp paired-end read metric, Amplicons are barcoded, multiplexed, and sequenced. Then the bioinformatic analysis can then be carried out.
References
- Castillo DJ, Rifkin RF, Cowan DA, et al. The healthy human blood microbiome: fact or fiction? Frontiers in cellular and infection microbiology. 2019, 9.
- Whittle E, Leonard MO, Harrison R, et al. Multi-method characterization of the human circulating microbiome. Frontiers in microbiology. 2019, 9.
- Li Q, Wang C, Tang C, et al. Identification and characterization of blood and neutrophil-associated microbiomes in patients with severe acute pancreatitis using next-generation sequencing. Frontiers in cellular and infection microbiology. 2018, 8.