Next-generation sequencing enables the detection of nearly all known pathogens simultaneously from various samples. The choice of approaches available depends on the priorities of sequencing and requires precision, quality, and cost tradeoffs. Most clinical and public health microbiology laboratories use short-read sequencing systems for routine sequencing, which create sequence fragments up to 1000 base-pair long. While microbial genomes are usually smaller and less complex than human genomes, techniques of next-generation sequencing are helpful for creating complete, highly precise genomes and figuring out plasmids, repeats, and other complex areas.
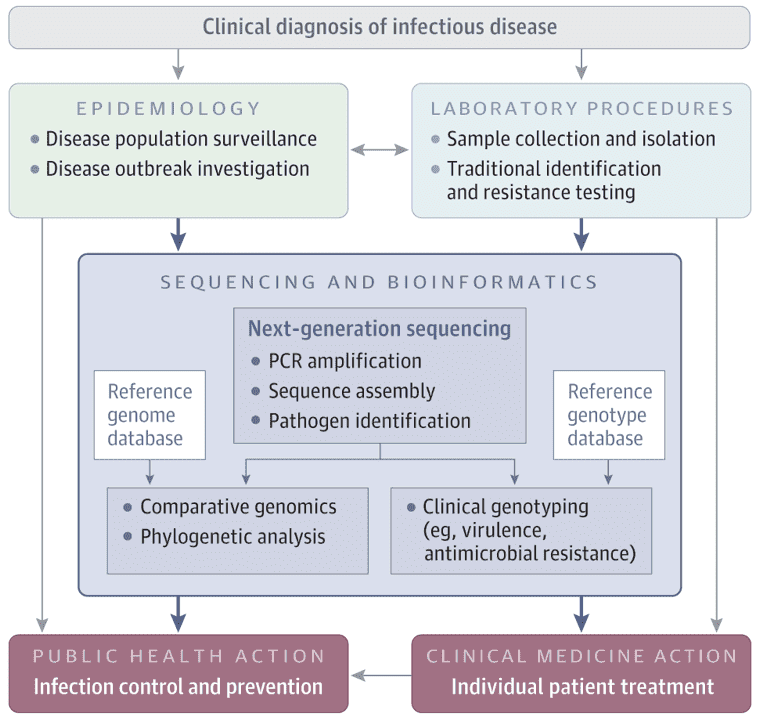 Figure 1. Workflow Converting genome sequence data from pathogens into credible data. (Gwinn, 2019)
Figure 1. Workflow Converting genome sequence data from pathogens into credible data. (Gwinn, 2019)
The translation of raw sequence data into actionable data is dynamic and computer-intensive (Figure 1). Usually, the first step is to compile shorter fragments into a complete sequence, either by mapping against a known reference genome or by using overlapping reads to arrange the de novo sequences. Many separate generalizations, such as pathogen recognition, high-resolution strain typing, and prediction of major phenotypic characteristics, are enabled by comparing the constructed genome with reference strains. Well-established and updated reference libraries are vital because microbial pathogens are rapidly emerging and bacteria can share plasmids through strains and organisms, also encoding virulence and antimicrobial resistance characteristics. As proof of replication, integrated genomes may be contrasted with others to search for phylogenetic clustering. Each phase involves various bioinformatics methods that must be harmonized into a coherent workflow: assembling, strain typing, phenotyping, and clustering.
Advantages of NGS on pathogen detection
In comparison to standard diagnostics of infectious diseases, such as those focused on culture, PCR, and serology, NGS-based diagnostics have some especially appealing features. Second, findings are less skewed than other approaches from NGS-based methods. Traditional diagnostics need some advanced knowledge of the handler, by default, while de novo NGS requires little to zero. To intensify a pathogen target by PCR, for example, allows the clinician to know what target pathogens to scan for. On the other hand, since NGS protocols typically use specific adapter sequences for DNA fragment priming and amplification, any DNA sequence present in the sample will potentially be sequenced without any previous knowledge of the organism. In addition, the unbiased nature of NGS enables co-infections to be identified that may normally be ignored by other assays.
NGS needs no prior evidence of a single organism's existence. Information about the genus and family of the organism is needed for other sequence-based assays, such as multi-locus sequence typing (MLST). A researcher could build an MLST panel and analyze the data online, but it would be important to sequence the resulting amplicons with Sanger and provide data about only a small collection of loci. A single NGS run, by comparison, can provide a far more information-rich data collection. In comparison, as opposed to using conventional assays, the time needed for actionable clinical evidence has been greatly decreased. An added benefit of NGS for the detection of infectious diseases is that non-culturable or fastidious pathogens may be identified by NGS.
References
- Gwinn M, MacCannell D, Armstrong GL. Next-generation sequencing of infectious pathogens. Jama. 2019, 321(9).
- Frey KG, Bishop-Lilly KA. Next-generation sequencing for pathogen detection and identification. Methods in microbiology. 2015, 42:525-54.