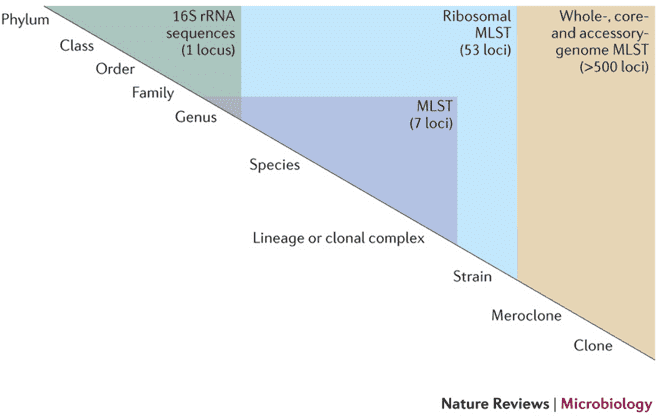
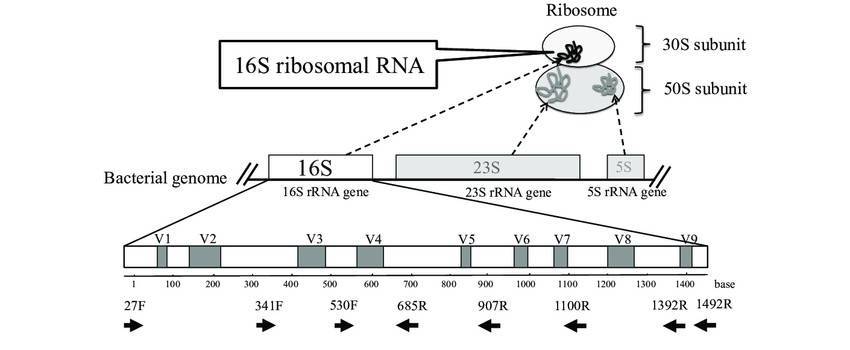
What is Microbial Diversity
Microbial diversity refers to the variation of many different types of microorganisms in a collective community and the relative abundance among them. Microbial diversity refers unequivocally to biological diversity at three levels: within species (genetic), species number (species) and community (ecological) diversity. In the past, the microorganisms of interest on the Earth were mainly those model bacteria and populations with direct economic value, while the most unknown populations that have been neglected also contain incalculable resources.
It is necessary to study microbial diversity for the following reasons (Tiedje et al., 1994): (1) The study of microorganisms in extreme environments not only enables us to understand and explore the strategies and limits of life, but also provides resources for the development and utilization of extraordinary substances; (2) Microorganisms can be used to monitor environmental changes, and their populations usually respond rapidly to environmental conditions, thus serving as good records of environmental changes in a region or history; (3) Microorganisms play an important role in the conservation and restoration biology of higher organisms. For example, mycorrhizal fungi can help plants successfully colonize the soil to achieve forest regeneration in the region. Human and animal microecology is a discipline based on the normal flora of human body and the harmony of human body. (4) Microbes serve as models for elucidating the principles of ecological and biological evolution. With the development of various microbial diversity analysis methods, we will be able to better study microbial diversity and apply it to life, production, environment and other aspects.
Methods for microbial diversity studies
Methods for studying microbial diversity have been developed over the years and can be divided into traditional research methods and modern molecular biology methods. The traditional microbial community analysis method is based on microbial isolation and pure culture, and the community structure is understood through microscopic observation and physiological and biochemical characteristics study of pure microorganisms, and several non-culture methods are gradually developed in response to the limitations of culture methods to study the types and quantities of microorganisms. These methods are broadly classified as biochemistry, physiology, etc.
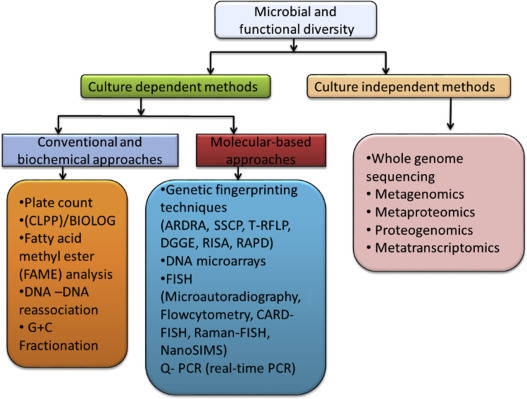
Traditional research methods
Traditional methods of microbial isolation and culture
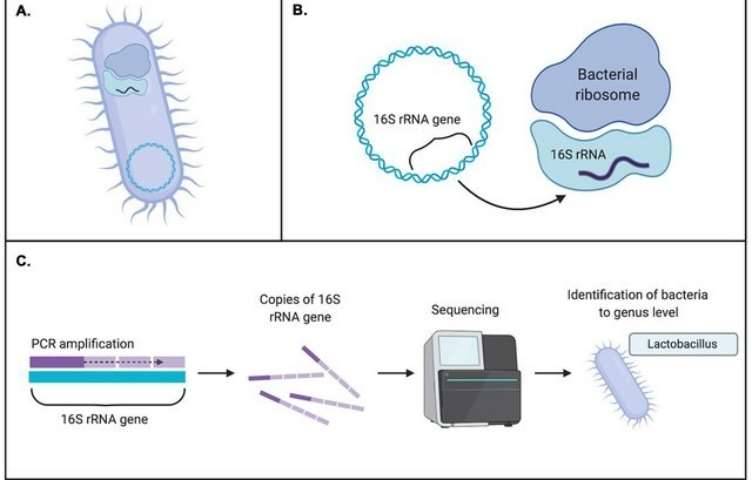
The process of obtaining only one kind or one strain of microorganism from a mixed microbial population is called microbial isolation and purification. The principle is to select suitable for microbial growth of nutrients, temperature and other conditions for environmental microbial culture. The general process of this method is as follows: gradient dilution of the sample to the appropriate concentration, coating on the corresponding solid medium, culture at the appropriate temperature, single colony purification, 16S rRNA gene amplification sequencing identification, storage. However, due to the complex microbial community structure and high species diversity in the environment, the method of pure isolation and enrichment culture is not only time-consuming and laborious, but also has fatal methodological defects: (1) the unculturability of a large number of microorganisms in nature makes it impossible for humans to culture all microorganisms in nature; (2) The separation and enrichment culture method has strong selectivity, so that the microorganisms obtained by culture can not reflect the real situation of microbial communities in the natural state in terms of variety and function (Rossell-mora et al., 2001).
Phospholipid fatty acid (PLFA)
PLFA exists in cell membranes, and because different microorganisms have different types and quantities of PLFA, it can be used as an indicator to understand the changes in the composition and number of microbial species in the natural environment. However, PLFA model can not give an actual microbial species composition, only a general map of community structure.
Biolog
Biolog microplate identification system is a system developed by Biolog Corporation to determine the utilization of 95 carbon sources by microorganisms and to identify chemotrophic bacteria based on this system. However, because different microorganisms have different utilization ability of the same carbon source, the difference of microbial metabolic fingerprints to different single carbon source cannot be simply summarized as the difference of microbial community quantity and structure, so the accuracy of Biolog method is limited to some extent. But the Biolog method is popular because it is fast and easy.
Modern molecular biology methods
As far as microbes are concerned, the cultivable ones are less in numbers, while uncultured or non-cultivable have just started showing up with the modern tools and techniques. These techniques help to increase our knowledge about the diversity of genetic resources and understand the distribution of organisms, the fundamental role of diversity, the ongoing interactions between various useful or pathogenic microbial communities, and a wide array of possibilities.
Modern molecular biology techniques mainly include denaturant gradient gel electrophoresis (DGGE)/temperature gradient gel electrophoresis (TGGE), restriction fragment length polymorphism (RFLP)/terminal restriction fragment length polymorphism (T-RFLP), Single Strand Conformation Polymorphism (SSCP), DNA microarrays, fluorescent in situ hybridization (FISH), metagenomic analysis, high-throughput technology etc.
Denaturant gradient gel electrophoresis/temperature gradient gel electrophoresis(DGGE/TGGE)
DGGE is used to analyze the diversity and relative richness of microorganisms by typing PCR products by electrophoresis. The principle of electrophoresis is used to separate DNA fragments by either a temperature or chemical gradient to denature the sample as it moves across an acrylamide gel. DNA segments which are same in length but have different base-pair sequences can be separated using these techniques. The separation is based on the difference in mobility of partially melted DNA molecules in acrylamide gels containing a linear gradient of DNA denaturants (urea and formamide). Hence, DNA sequences having a difference in only one base-pair can be separated by DGGE. TGGE employs the same principle as DGGE, but in this method, the gradient is temperature rather than chemical denaturants. DGGE was originally developed as a technique to study mutational sites in DNA sequences. In 1993, Muyzer et al. (1993) first applied DGGE to the study of microbial genetic diversity. PCR-DGGE a culture-independent approach that has been used to analyze microbial community structure across different fields, such as food microbiology, oral microbiology, soil microorganisms, environmental microbiology, and other areas (Sidira et al., 2014; de Paula et al., 2014; Jonathan et al., 2013; Zhong et al., 2014).
Restriction fragment length polymorphism/ terminal restriction fragment length polymorphism (RFLP/T-RFLP)
Another approach to study microbial diversity is restriction fragment length polymorphism (RFLP). PCR-RFLP is a combination of PCR technology, RFLP analysis and electrophoresis. First, the target DNA fragment to be detected is replicated and amplified, and then the amplified product is digested by DNA restriction endonuclease. Finally, it is analyzed by electrophoresis whether the target DNA fragment is cut or not. Another variant of this technique is terminal RFLP that addresses some of the limitations of RFLP by providing an alternate method for rapid analysis of microbial community diversity in different environments. Kanokratana P et al. (2004) used RFLP to study prokaryotic diversity in the Bor Khlueng hot springs, Thailand; Everroad R C et al. (2012) used T-RFLP analysis to study the effect of temperature on microbial diversity in Nakabusa hot springs, Japan, in order to better understand the biogeography and relationship between temperature and community structure within microbial mats. Based on the principal of RFLP, there are other techniques that were used sporadically. They are RISA and the automated version called ARISA. Here, the intergenic spacer region between the 16S and 23S ribosomal subunits is amplified by PCR and separated on a polyacrylamide gel under denaturing conditions. This is found to have differentiating potential for bacterial strains and closely related species (Fisher and Triplett, 1999).
Single strand conformation polymorphism (SSCP)
Single strand conformation polymorphism (SSCP) also relies on electrophoretic separation based on differences in DNA sequences and allows differentiation of DNA molecules having the same length but different nucleotide sequences. This technique was originally developed to detect known or novel polymorphisms or point mutations in DNA (Peters et al., 2000). In this method, single-stranded DNA separation on polyacrylamide gel was based on differences in mobility resulted from their folded secondary structure (Heteroduplex) (Lee et al., 1996). As formation of folded secondary structure or heteroduplex and hence mobility is dependent on the DNA. SSCP also suffers from the limitations as like DGGE, and hence the same DNA sequence can produce multiple bands on the gel. However, it does not require gradient gel and has been used for diversity studies (Stach et al., 2001).
Microarrays
Gene microarrays have been used to analyze the composition and diversity of microorganisms in samples for nearly 20 years, and have undergone several generations of improvement. The commonly used microarrays include PhyloChip (used to identify microorganisms and their phylogenetic relationships and analyze the diversity of microorganisms) and Functional GeoChip (used to study the diversity of functional genes and the activity of functional microorganisms). Microarray is a chip filled with probes (known short sequences for hybridization with sample DNA), which provide phylogenetic information or functional property information of organisms, or both. When the sequence in the sample (the fluorescence-labeled PCR product of the target fragment of DNA or RNA of pure bacteria and environmental samples or the fluorescence-labeled PCR product of random primers) hybridizes with the probe, the relative fluorescence ratio of the sequence matched with the probe can be calculated, so as to obtain the diversity and relative richness information of microorganisms. Gene microarray method is especially suitable for identifying the differences of representative microorganisms or microbial communities between different times, places and treatments. In addition, PhyloChip can also quantify the changes of related functional genes such as C, N, S and P cycles, degradation of organic pollutants and stress response. Aronson et al. (2013) used the third generation PhyloChip(16S rRNA gene microarray, which can provide about 60,000 different OTU information) to study the microorganisms related to methane cycle in pine forest soil under different experimental conditions.
Fluorescent in situ hybridization (FISH)
FISH is one of the most popular methods for DNA hybridization. Nucleic acid hybridization using specific probes is an important qualitative and quantitative tool in molecular bacterial ecology (Clegg et al., 2000). These hybridization techniques can be done on extracted DNA or RNA, or in situ. The sample is lysed to release all nucleic acids. Dot-blot hybridization with specific and universal oligonucleotide primers is used to quantify rRNA sequences of interest relative to total rRNA. The relative abundance may represent changes in the abundance in the population or changes in the activity and hence the amount of rRNA content (Theron and Cloete, 2000). Spatial distribution of bacterial communities in different environments such as biofilms can be determined using FISH (Schramm et al., 1996).
Metagenomic analysis
Metagenomics is defined as the functional and sequence-based analysis of the collective microbial genomes that are contained in an environmental sample (Zeyaullah et al., 2009). This contains all the genomes from coexisting microbes-called microbial communities. This is further subjected to the sequencing to know about the species composition in the sample. This provides a comprehensive view of the genetic diversity, species composition, evolution, and interactions with the environment of natural microbial communities (Simon and Daniel, 2011).
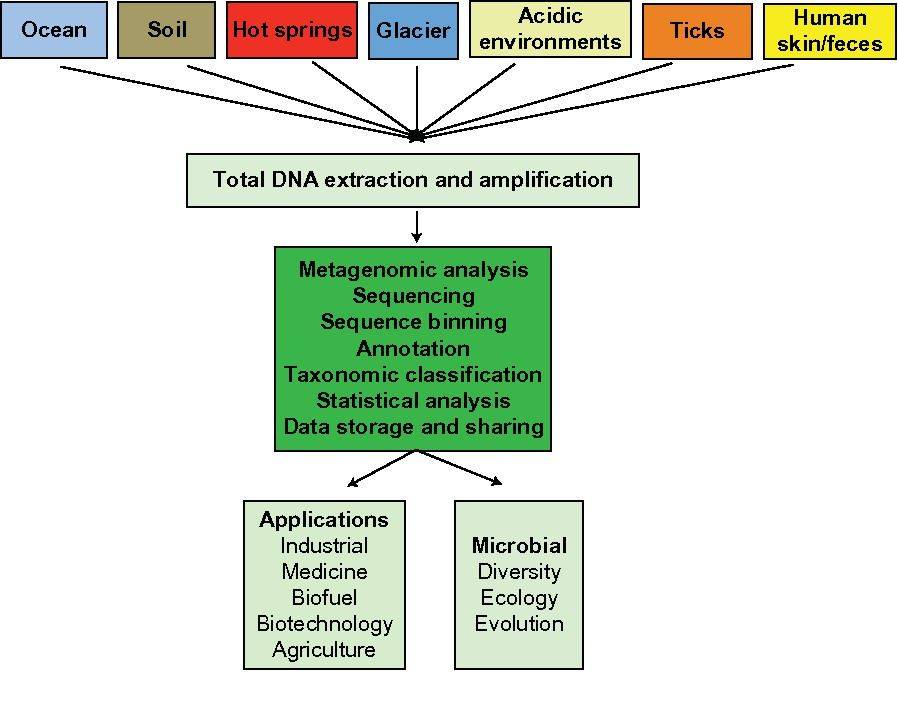 Overview of metagenomic analysis. (Girish Neelakanta et al,.2013)
Overview of metagenomic analysis. (Girish Neelakanta et al,.2013)
High-throughput technology
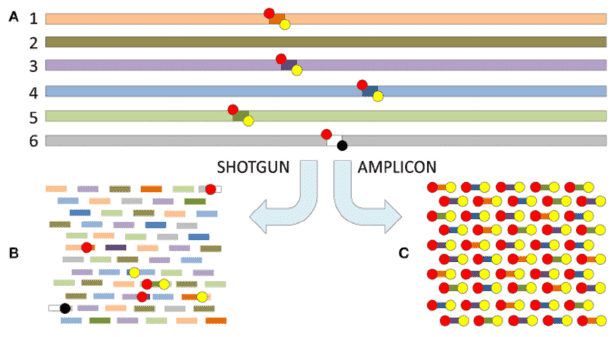
Sanger sequencing has been used for decades, and the sequencing quality is high, with a sequence length of 750—1000 bp. This method can't measure the mixed sequence, so it is necessary to establish a clone library first, that is, transfer the target sequence to the host cell for culture to form a single strain, and then sequence the target sequence in the single strain. It is time-consuming and expensive to analyze environmental samples with high microbial diversity.
On the basis of Sanger sequencing, high-throughput sequencing, also called next-generation sequencing (NGS), was developed. This method used PCR products of microbial target genes as samples to sequence, and tens of thousands to millions of sequences were obtained in one reaction, which greatly improved the breadth and depth of sequencing. At present, amplicon sequencing is commonly used methods for microbial diversity analysis in environmental samples, namely, 16S/18S/ITS sequencing. These high-throughput sequencing methods can identify the sequences of different samples by labeling the primers of each sample, and can simultaneously analyze multiple environmental samples and obtain a large number of sequences.
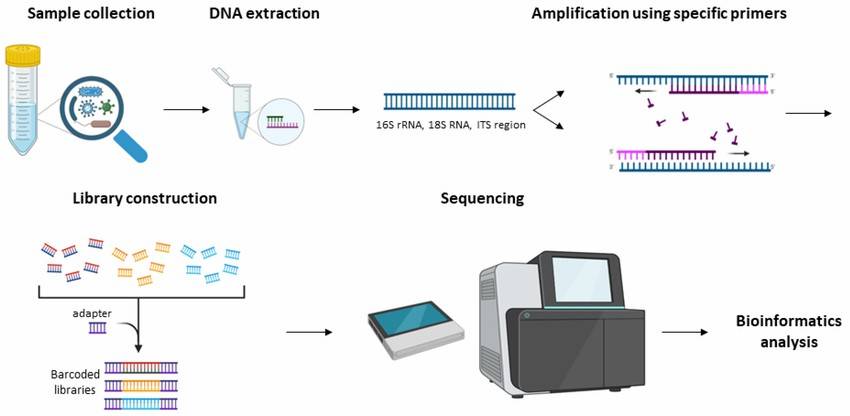 Demonstration of the workflow for the 16S rRNA and ITS amplicon sequencing.(Konstantina Athanasopoulou et al,. 2023)
Demonstration of the workflow for the 16S rRNA and ITS amplicon sequencing.(Konstantina Athanasopoulou et al,. 2023)
Application of microbial diversity research
Environmental issues
The study of microbial diversity is helpful to increase our understanding of environmental pollution and provide treatment programs. In contaminated sites, microbial communities are the key driving force behind the biological activities and treatment of pollutants. Effective and reliable bioremediation strategies require periodic measurement of physicochemical parameters and a complete understanding of the functioning of degrading microbial communities, including the relationship between their structure (species composition) and their function (catabolic properties). Soil bacteria play a major role in ecological and biodegradable function processes in contaminated soils. Soils contaminated with oil have shown more diversity of bacteria and a shift based on organic matter and exposure time compared to clean soils when analyzed using NGS. This is a useful information for bioremediation and finding the role of new communities in cleaning oil spills (Peng et al., 2015; Joshi et al., 2014). Bioindicators of sewage contamination in a water environment is another field of interest to public health which can be handled using NGS (Tan et al., 2015). Jiang et al (2021) used Illumina MiSeq sequencing technology to investigate the seasonal relationship between bacterial and fungal microbial diversity and environmental factors in a large drinking water reservoir, this work is crucial to better under-stand microbial characteristics and choose an appropriate management strategy for the maintenance of reservoirs.
Medicine
Microbial communities are critical in the human body, with the gut having the highest microbial diversity, and hence gut microbiome studies have emerged as a new branch of exploration and intervention. Similarly individual studies on the oral microbiome (Yamashita et al., 2017), lung microbiome (Gao et al., 2022), skin microbiome (Sandhu et al., 2019), etc., are being explored using the power and resolution of NGS. Even microbial roles are now being predicted in some kind of cancers (Faden, 2016). Microbial diversity analysis using NGS has also contributed to new drug discovery efforts. The genome mining with NGS has helped in identifying new natural products and their biosynthetic pathways (Adu-Oppong et al., 2017).
Agriculture
Agriculture is one such man-made system that is influenced by the microbial diversity and also influences the diversity in turn. Soil microorganisms substantially contribute to the resistance and resilience of agroecosystems to abiotic disturbance and stress. Hence, the richness of species of plants and microbes determines the productivity of the land. Adhikari et al. used NGS to study the microbial structure and diversity in watermelon cultivated soils from different regions, environmental factors play a key role in bacterial community and diversity, and they will further analyze the correlation between soil physical and chemical parameters and dominant bacterial communities to observe their interactions (Adhikari et al., 2021). Bevivino and Dalmastri (2017) used NGS to illustrate the effects of different agricultural management practices on the structure and function of soil microbial communities. They also described the effects of tillage and nitrogen fertilizer on soil bacterial community structure.
Industrial activities
The study of microbial diversity is also applied to industrial activities. Joshi et al (2016) have tried to provide quick pragmatic evidence of aquatic microbial diversity and focus on some of their industrial important enzymes. New and better strains for fermentation industry are being explored from a previously uncultivable lot of microbes which are now identified and characterized by the use of NGS or functional metagenomics. By studying the microbial diversity of kraft paper mill sludge, Ghribi et al. (2016) demonstrated that the strains found in the sludge represent a source of potential novel enzymes for industrial applications and bioremediation. Microbes with new functions and industrial applications have been identified from oceans under Global Ocean Sampling Expedition (Williamson et al., 2008). In addition, Wei et al (2022) found that NGS could be used to study the microbial diversity of industrial wastewater to trace the source of wastewater.
Applications in other activities
Microbial diversity may also be useful in biosurveillance and forensics. In some cases, human microbial signatures have been used to match individuals to objects they have interacted with, including computer keyboards (Fierer et al., 2010). Work on the microbiome of multiple home surfaces has shown that the microbial signature of a family can be highly predictive of the microbiome of that family’s home and that individuals within a home can be differentiated (Lax et al., 2014). A study has revealed that after the death, the microbiome of animal hosts changes dramatically, but in a predictable manner (Metcalf et al., 2013) which can assist in tracing the time and direction of the events.
Conclusions and prospects
Microbial diversity in natural environments is extensive. Methods for studying diversity vary and diversity can be studied at different levels, i.e. at global, community and population levels. The molecular perspective gives us more than just a glimpse of the evolutionary past; it also brings a new future to the discipline of microbial ecology. The development and progress of molecular biology methods promote the study of microbial diversity, but different methods have their inherent limitations, and the selection and application of methods should be based on the specific research situation. However, these methods also have some problems. For example, high-throughput sequencing usually produces hundreds of bases or shorter sequences, so it is difficult to form a complete gene. Even if all these bases are obtained, it is difficult to accurately map from sequence to function because the functions of many genes are still unclear. In addition, due to the problems of selection and PCR itself, microorganisms may be misunderstood. The obtained information can be easily amplified by PCRs, and most of the increased populations are dominant populations. But generally speaking, in recent years, the breadth, depth and resolution of molecular biology methods have been greatly improved, showing unprecedented advantages and application prospects. The application of microbial diversity analysis in all aspects of life has promoted our understanding of microbial diversity, including from different parts of the human body and different parts of the earth. In the future, more research may use microbial diversity analysis to find specific microorganisms for production and life. Monitoring microbial diversity is very important to analyze the succession law of community and understand the dynamic characteristics of population. A comprehensive understanding of microbial diversity is helpful to reveal the interaction between microorganisms and the environment. The study of microbial diversity has promoted the development of environmental science and ecology, and will continue to make important contributions to improving human health, increasing crop yield and developing clean energy.
References
- Adhikari M, Kim S W, Kim H S, et al., 2021. Bacterial Community and Diversity from the Watermelon Cultivated Soils through Next Generation Sequencing Approach. The Plant Pathology Journal, 37(6): 521.
- Adu-Oppong, B., Gasparrini, A.J., Dantas, G., 2017. Genomic and functional techniques to mine the microbiome for novel antimicrobials and antimicrobial resistance genes. Ann. N. Y. Acad. Sci. 1388 (1), 42-58.
- Aronson E L, Dubinsky E A, Helliker B R.,2013. Effects of nitrogen addition on soil microbial diversity and methane cycling capacity depend on drainage conditions in a pine forest soi. Soil Biology and Biochemistry, 62: 119-128.
- Bevivino, A., Dalmastri, C., 2017. Impact of agricultural land management on soil bacterial community: a case study in the Mediterranean Area. In: Lukac, M., Grenni, P., Gamboni, M. (Eds.), Sustainability in Plant and Crop Protection. Springer, Cham, Soil Biological Communities and Ecosystem Resilience.
- Clegg, C.D., Ritz, K. and Griffiths, B.S., 2000. %G+C profiling and cross hybridisation of microbial DNA reveals great variation in below-ground community structure in UK upland grasslands. Applied Soil Ecology 14: 125– 134.
- de Paula, V.A., de Carvalho Ferreira, D., Cavalcante, F.S., do Carmo, F.L., Rosado, A.S., Primo, L.G., et al., 2014. Clinical signs and bacterial communities of deciduous necrotic root canals detected by PCR-DGGE analysis: research association. Arch. Oral. Biol. 59 (8), 848-854.
- Everroad R C, Otaki H, Matsuura K, et al., 2012. Diversification of bacterial community composition along a temperature gradient at a thermal spring. Microbes and environments, 27(4): 374-381.
- Faden, A.A., 2016. The potential role of microbes in oncogenesis with particular emphasis on oral cancer. Saudi. Med. J. 37 (6), 607-612.
- Fierer, N., Lauber, C.L., Zhou, N., et al., 2010. Forensic identification using skin bacterial communities. Proc. Natl. Acad. Sci. U.S.A. 107 (14), 6477-6481.
- Fisher, M.M., Triplett, E.W., 1999. Automated approach for ribosomal intergenic spacer analysis of microbial diversity and its application to freshwater bacterial communities. Appl. Environ. Microbiol. 65 (10), 4630-4636.
- Gao xiaohui, Cai Z, Guo Q, et al., 2022. Insights into the unique lung microbiota profile of pulmonary tuberculosis patients using metagenomic next-generation sequencing. Microbiology Spectrum, 10(1): e01901-21.
- Ghribi M, Meddeb-Mouelhi F, Beauregard M.,2016. Microbial diversity in various types of paper mill sludge: identification of enzyme activities with potential industrial applications. Springerplus, 5(1): 1-14.
- Jiang T, Sun S, Chen Y, et al. 2021., Microbial diversity characteristics and the influence of environmental factors in a large drinking-water source. Science of The Total Environment, 769: 144698.
- Jonathan, L., Richard, V., Louise, D., 2013. A new framework to accurately quantify soil bacterial community diversity from DGGE. Microb. Ecol. 66 (3), 647-658.
- Joshi, M.N., Dhebar, S.V., Bhargava, P., et al., 2014. Metagenomic approach for understanding microbial population from petroleum muck. Genome Announc. 2 (3).
- Joshi P, Pande V, Joshi P., 2016. Microbial diversity of aquatic ecosystem and its industrial potential. J. Bacteriol. Mycol. Open Access, 3: 177-179.
- Kanokratana P, Chanapan S, Pootanakit K, et al., 2004. Diversity and abundance of Bacteria and Archaea in the Bor Khlueng hot spring in Thailand. Journal of Basic Microbiology: An International Journal on Biochemistry, Physiology, Genetics, Morphology, and Ecology of Microorganisms, 44(6): 430-444.
- Lax, S., Smith, D.P., Hampton-Marcell, J., et al., 2014. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 345 (6200), 1048-1052.
- Lee, D.H., Zo, Y.G. and Kim, S.J., 1996. Nonradioactive method to study genetic profiles of natural bacterial communities by PCR single strand conformation polymorphism. Applied and Environmental Microbiology 62: 3112– 3120.
- Metcalf, J.L., Wegener Parfrey, L., Gonzalez, A., et al., 2013. A microbial clock provides an accurate estimate of the postmortem interval in a mouse model system. eLife 2, e01104.
- Muyzer G, De Waal E C, Uitterlinden A G., 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Applied and environmental microbiology, 59(3): 695-700.
- Peng, M., Zi, X., Wang, Q., 2015. Bacterial community diversity of oil-contaminated soils assessed by high throughput sequencing of 16S rRNA genes. Int. J. Environ. Res. Public. Health 12 (10), 12002-12015.
- Peters, S., Koschinsky, S., Schwieger, F. and Tebbe, C.C., 2000. Succession of microbial communities during hot composting as detected by PCR-single-strand-conformation polymorphism based genetic profiles of small-subunit rRNA genes. Applied and Environmental Microbiology 66: 930– 936.
- Rossell-mora R, Amann R., 2001. The species concept for prokaryotes. FEMS Micro Rev, 25(1): 39-67.
- Sandhu S S, Pourang A, Sivamani R K., 2019. A review of next generation sequencing technologies used in the evaluation of the skin microbiome: what a time to be alive. Dermatology online journal, 25(7).
- Schramm, A., Larsen, L.H., Revsbech, N.P., et al., 1996. Structure and function of a nitrifying biofilm as determined by in situ hybridization and the use of microelectrodes. Applied and Environmental Microbiology 62: 4641.
- Sidira, M., Karapetsas, A., Galanis, A., et al., 2014. Effective survival of immobilized Lactobacillus casei during ripening and heat treatment of probiotic dry-fermented sausages and investigation of the microbial dynamics. Meat. Sci. 96 (2 Pt A), 948-955.
- Simon, C., Daniel, R., 2011. Metagenomic analyses: past and future trends. Appl. Environ. Microbiol. 77 (4), 1153-1161.
- Stach, J.E., Bathe, S., Clapp, J.P., Burns, R.G., 2001. PCR-SSCP comparison of 16S rDNA sequence diversity in soil DNA obtained using different isolation and purification methods. FEMS Microbiol. Ecol. 36 (2-3), 139-151
- Tan, B., Ng, C., Nshimyimana, J.P., et al., 2015. Next-generation sequencing (NGS) for assessment of microbial water quality: current progress, challenges, and future opportunities. Front. Microbiol. 6, 1027.
- Theron, J. and Cloete, T.E., 2000. Molecular techniques for determining microbial diversity and community structure in natural environments. Critical Reviews in Microbiology 26: 37– 57.
- Tiedje J M., 1994. Microbial diversity: Of value to whom. ASM News, 60: 524-525.
- Williamson, S.J., Rusch, D.B., Yooseph, S., et al., 2008. The Sorcerer II Global Ocean Sampling expedition: metagenomic characterization of viruses within aquatic microbial samples. PLoS ONE 3 (1), e1456.
- Wei Y, Li Y, Wang Y, et al. 2022. The microbial diversity in industrial effluents makes high-throughput sequencing-based source tracking of the effluents possible. Environmental Research, 212: 113640.
- Yamashita Y, Takeshita T., 2017. The oral microbiome and human health. Journal of oral science, 59(2): 201-206.
- Zeyaullah, M., Kamli, M.R., Islam, B., et al., 2009. Metagenomics an advanced approach for noncultivabl micro-organisms. Biotechnol. Mol. Biol. Rev. 4 (3), 049-054.
- Zhong, X., Rimet, F., Jacquet, S., 2014. Seasonal variations in PCR-DGGE fingerprinted viruses infecting phytoplankton in large and deep perialpin lakes. Ecol. Res. 29, 271-287.