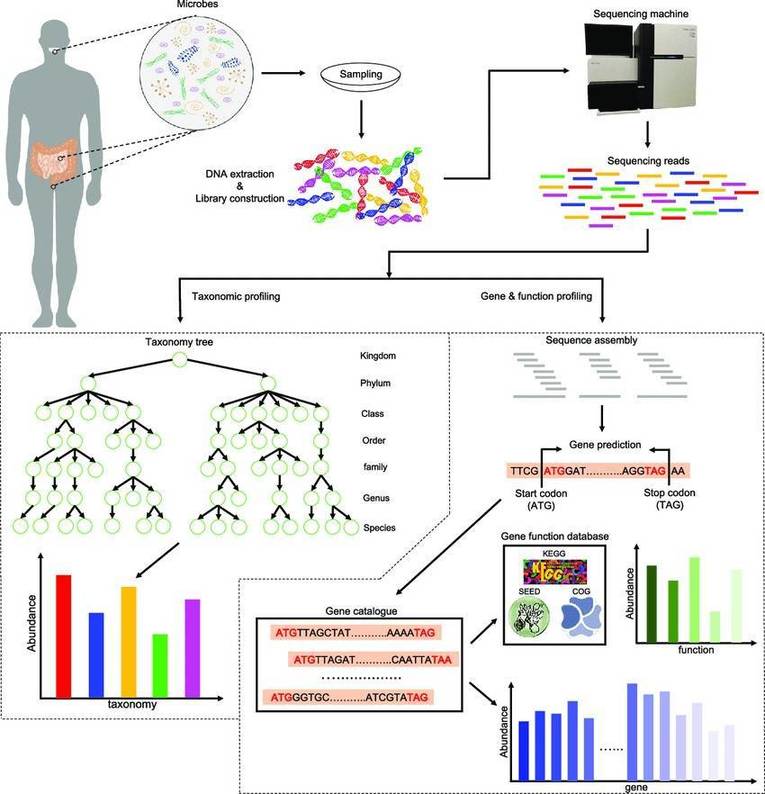
In the fields of microbial ecology, environmental science, and biodiversity research, Metabarcoding and Metagenomics have become the core tools to analyze complex biological communities with their unique advantages. However, there are significant differences in research objectives, technical principles, and application scenarios, and they also face inherent limitations.
This paper will analyze the changes brought by the technology selection framework, inherent limitations and deviations, quantitative accuracy, collaborative application mode, and long reading and long sequencing, and provide a comprehensive reference for technology selection and scheme design of related research.
Choosing the Right Tool: A Decision Framework
It is the key to ensuring the research efficiency and the reliability of the results to accurately select macro-bar code or macro-genomics technology before conducting the research on biological communities. The application scenarios of the two can be divided into three dimensions: research objectives, cost constraints, and sample characteristics, forming a clear decision-making basis.
Preference Given to DNA Metabarcoding
When the research focuses on issues related to species classification, or is limited by cost and sample quality, the macro barcode technology shows significant advantages, and the specific application scenarios include:
- Biodiversity survey: In the general survey of species composition of ecosystems, such as the monitoring of microbial community diversity in forest soil, the survey of plankton species in fresh water, etc., DNA Metabarcoding can quickly obtain the classification information of species in samples by amplifying and sequencing specific bar code genes, and detect hundreds to thousands of taxonomic units at one time, and the cost is only 1/5-1/10 of that of macro-genomics, which can effectively cover a large number of samples and meet the needs of large-scale and multi-site diversity survey.
- Biological monitoring: In scenarios of monitoring the impact of environmental pollutants on biological communities, early detection of invasive species, etc., a metabarcoding can quickly evaluate the changes in community structure.
- Analysis of degraded DNA samples: For environmental DNA, fossil DNA, or improperly preserved samples, their DNA is usually seriously fragmented and low in concentration. Short-segment barcode genes (usually 200-500bp) targeted by metabarcoding are more likely to obtain effective sequences through PCR amplification, while metagenomics needs long, complete DNA fragments to construct a library, which is difficult to apply to such samples.
Metagenome as the First Choice
When the research goal involves functional gene analysis, special biological group research, or genome reconstruction, metagenomics technology becomes an inevitable choice, and the specific application scenarios include:
- Functional mechanism research: In the fields of microbial metabolic pathway analysis and environmental pollutant degradation mechanism research, metagenomics can mine genes related to specific functions by sequencing all DNA in samples.
- Antibiotic resistance gene research: In the investigation of antibiotic resistance genes (ARGs) in clinical environment or environmental media (such as sewage and soil), metagenomics can comprehensively detect all types of ARGs in samples, including known and unknown resistance genes, and analyze their carrying vectors (such as plasmids and transposons) to evaluate the risk of the spread of resistance genes.
- Discovery of new genes: Metagenomics does not need to rely on known gene sequences, but can find new genes that have not been reported by splicing and annotating sequence data.
- Virus/phage research: Viruses and phages have no common barcode genes, and their genomes are diverse, so macro barcodes cannot effectively amplify and classify them. Metagenomics can directly sequence all viral DNA/RNA in the sample, obtain the viral genome sequence through genome assembly, and then analyze the virus species, host range, and pathogenic mechanism.
- Genome reconstruction: In the genome analysis of the target microorganism, metagenomics can obtain the nearly complete genome (MAGs) of an uncultured microorganism by assembling the sequencing data.
 Proposed workflow of a clinical microbiology laboratory using a CMg approach to identify putative pathogens (Forbes et al., 2018)
Proposed workflow of a clinical microbiology laboratory using a CMg approach to identify putative pathogens (Forbes et al., 2018)
| Explore Related Research Services | Resource |
|---|---|
| Microbial DNA Metabarcoding | Five Dimensions for Metabarcoding and Metagenomics Comparison |
| Metagenomics | Metagenomics Sequencing Overview |
Inherent Limitations and Biases of Each Approach
Although DNA metabarcoding code and metagenomics technology are widely used in research, due to the influence of technical principles and experimental procedures, there are inevitable inherent limitations and deviations, which directly affect the accuracy of results and the reliability of interpretation.
A. Limitations and deviations of the Metabarcoding
a) DNA Metabarcoding depends on PCR amplification and specific bar code genes, and its limitations mainly come from the amplification process, gene selection, and reference database, which are as follows:
i. Deviation between PCR and primers: In the process of PCR amplification, there are differences in the binding efficiency between template DNA and primers of different species, which leads to the amplification efficiency of some species being much higher than that of other species, thus making the sequence reading length of this species overestimated, while the species with low amplification efficiency (such as some rare microorganisms) are underestimated or even omitted, which can not truly reflect the relative abundance of species in the sample.
ii. Limited to the organisms covered by the target barcode gene: Metabarcoding can only detect the organisms carrying the target barcode gene, but can't identify the organisms lacking the gene or whose gene sequence is too variable.
iii. Lack of direct functional information: Metabarcoding can only provide species classification information, but cannot relate to the functional characteristics of organisms.
iv. Completely dependent on the reference database: The accuracy of species annotation depends entirely on the integrity and accuracy of the sequence in the reference database. At present, a large number of species (especially rare species and undiscovered new species) are still missing in public reference databases (such as GenBank and Silva), which makes these species unable to be annotated in macro bar code results (i.e., "unidentified taxon"), accounting for 10%-40%. In addition, some sequences in the database have classification errors or incomplete annotation information (for example, they are only labeled at the genus level, but not at the species level), which further affects the accuracy of species identification.
B. Limitations and deviations of metagenomics
b) Although metagenomics can obtain all the DNA information in the sample, it is also obviously limited by the cost, sample quality, and data analysis methods, which are as follows:
i. The cost and computational burden are significantly higher: the sequencing depth of metagenomics usually needs to reach 10-100 bp/sample, which is much higher than that of macro bar code (usually 0.5-2 bp/sample), and the sequencing cost is about 5-20 times that of macro bar code. At the same time, the assembly, annotation and analysis of marine sequence data (usually millions to tens of millions of sequences) require high-performance computing resources (such as the memory in the server needs more than 64G, and the analysis period is as long as several weeks), and it needs professional bioinformatics personnel to operate, which is difficult for small laboratories or research projects with limited funds.
ii. The quality and quantity of DNA are high: the construction of a metagenomics library needs complete and high-quality DNA (the fragment length is usually ≥1kb), and the DNA concentration needs to reach a certain threshold (usually ≥100ng/μL). For low-concentration DNA (< 10 ng/μL) or severely degraded DNA (fragment length < 500bp) in environmental samples (such as soil and water), it is difficult to construct a high-quality library, resulting in a low proportion of effective sequences in sequencing data, and even the sequencing cannot be completed.
iii. Low-abundance genome assembly is difficult: Genome assembly in metagenomics depends on sufficient sequence coverage. For microorganisms with an abundance of less than 0.1% in samples, the coverage of genome sequences is usually insufficient, and it is difficult to assemble complete genome fragments, resulting in the information of these low-abundance microorganisms being omitted.
iv. There is uncertainty in function prediction: metagenomics predicts biological function by detecting the existence of functional genes, but the existence of genes is not the same as gene expression.
 Simplified workflow of current DNA-based methods for analysis of (micro) organisms within environmental samples, using honey as an example (Vuong et al., 2024)
Simplified workflow of current DNA-based methods for analysis of (micro) organisms within environmental samples, using honey as an example (Vuong et al., 2024)
The Quantitative Dilemma: Which is More Accurate
In the study of biological communities, it is the key to analyzing the dynamic changes of community structure and evaluating the interaction between species to accurately quantify the relative abundance or genome abundance of species in samples. However, both metabarcoding and metagenomics have defects in quantitative accuracy, forming their own quantitative dilemmas, and the difference between them is mainly due to the influence of technical principles on abundance assessment.
DNA Metabarcoding: Essentially Semi-quantitative Technology
Metabarcoding infers the relative abundance of species by the sequence reading length obtained by sequencing, but its quantitative result is only semi-quantitative due to the variation of PCR amplification deviation and gene copy number, which can not accurately reflect the true abundance of species. The specific problems include:
- The deviation of PCR amplification leads to the distortion of abundance: The combination efficiency of template DNA and primers of different species is different, and the species with high amplification efficiency (such as some Gram-negative bacteria) will produce more PCR products, and then get more sequence readings in the sequencing results, which leads to the overestimation of its abundance. However, species with low amplification efficiency (such as Gram-positive bacteria) will be underestimated because of less PCR products and low sequence reading length.
- The variation of gene copy number interferes with abundance calculation: the copy number of the same barcode gene (such as the 16S rRNA gene) in different species is significantly different in the genome. For example, the copy number of the 16S rRNA gene of bacteria can range from 1 (such as mycoplasma) to 15 (such as Bacillus subtilis). Species with high copy number will produce more barcode gene templates and gain more sequence reading length after PCR amplification, which leads to overestimation of their abundance.
Therefore, the quantitative results of macro bar code can only be used to compare the relative change trend of species abundance in different samples, but they can not accurately give the actual cell number or true relative abundance of species.
Metagenomics: A Better Choice of Relative Genome Abundance
Metagenomics directly sequences all the DNA in the sample without PCR amplification, and evaluates the relative genome abundance of species by the ratio of genome coverage or sequence reading length to genome size, which avoids the PCR amplification deviation and has better quantitative accuracy than macro bar code, but it is still unable to achieve absolute quantification due to the difference in genome size. The specific features include:
- Avoid PCR bias, and the relative abundance is closer to the truth: Metagenomics randomly interrupts and sequences the DNA in the sample without PCR amplification, so there is no abundance distortion caused by the difference in primer binding efficiency.
- Genome size has become a new interference factor: The quantitative results of metagenomics reflect the relative genome abundance, not the relative cell abundance. Because the genome size of different species is significantly different (for example, the genome size of bacteria is usually 1-10 Mb, and that of fungi is 10-100 Mb), under the condition of the same number of cells, the species with large genome will contribute more DNA, and then get more sequence readings in the sequencing results, which leads to its relative genome abundance being overestimated.
Therefore, although the quantitative results of metagenomics are more accurate than that of metabarcoding, which can better reflect the relative genomic abundance of species, when it is necessary to evaluate the relative cell abundance or absolute cell number, it still needs to be corrected by combining with other technologies (such as flow cytometry and qPCR), and it is impossible to achieve completely accurate quantification alone.
 Case examples illustrating different scenarios for MAG-V9 OTU match scenarios, results for the "rho" proportionality metrics (Zavadska et al., 2024)
Case examples illustrating different scenarios for MAG-V9 OTU match scenarios, results for the "rho" proportionality metrics (Zavadska et al., 2024)
Synergy in Modern Studies: The Hierarchical Pipeline
In ecological research, DNA metabarcoding and metagenomics have changed from independent applications to collaborative integration, and a hierarchical research paradigm with resource optimization and precise focus as the core has been constructed. This paradigm adopts the progressive strategy of "initial screening-deep analysis" and becomes the mainstream technical framework of complex ecological research.
The hierarchical research process follows the logic of "Qualcomm screening-in-depth analysis": the key samples and core groups are quickly locked by metabarcoding, and the functional mechanism of communities is revealed by macro-genomics. Its implementation is divided into three stages:
- Large-scale sample screening: For dozens to hundreds of samples (such as soils with different pollution gradients and water bodies in different seasons), the target genes (16S rRNA of bacteria and COI of eukaryotes) are sequenced by macro bar code technology, and the basic information of community structure can be obtained, and the different sample groups and core groups can be quickly identified, and the analysis of 100-level samples can be completed within 1-2 weeks.
- Selection of key samples: According to the preliminary screening results and research objectives, 10-20 representative samples were screened to avoid ineffective investment in metagenomics.
- In-depth analysis of metagenomics: Sequencing key samples to obtain metagenome assembly genomes (MAGs), mining functional genes, and constructing a species-function-environment factor correlation model to realize a closed-loop demonstration of structure-function.
This process has obvious advantages: At the resource level, the research cost will be reduced by 50%-70%. In terms of research depth, targeted function analysis improves efficiency. In terms of reliability, the two confirmed each other.
At present, it has been widely used in soil remediation, water treatment, intestinal microorganisms, and other research fields, and has become the core technical path to connect community structure and functional mechanism research.
 Abundance of two marine microbes illustrated by three hypothetical scenarios (Brennan et al., 2023)
Abundance of two marine microbes illustrated by three hypothetical scenarios (Brennan et al., 2023)
The Future with Long-Read Sequencing: Blurring the Lines
The development of long-read sequencing technologies, such as Oxford nanopore and PacBio single-molecule real-time sequencing, has broken the traditional technical barriers between macro barcode and metagenomics. With the sequencing reading length of several kilobytes to tens of kilobytes, it can cover both barcode genes and functional genes at the same time, realize the synchronous acquisition of single reading length of species classification and functional information, and effectively solve the problem that the structure and function of traditional technology are out of touch.
This technology has achieved breakthroughs in three dimensions:
- First, it has achieved single reading long classification-function dual information acquisition, which has overcome the defects of traditional macro bar codes that have no functional correlation and the fuzzy species-function correspondence caused by metagenomics splicing. For example, in bacterial research, long-read sequences can cover both 16S rRNA and nifH genes, and directly establish the species-function correspondence.
- Second, the high-resolution analysis of complex communities is realized, which makes up for the deficiency that traditional short reading length makes it difficult to distinguish related species. For example, in the study of human intestinal Escherichia coli, pathogenic and symbiotic strains are accurately distinguished by covering virulence genes with long fragments.
- Thirdly, simplify the experiment and analysis process, avoid the primer deviation of traditional macro bar code and the complex splicing problem of metagenomics, and shorten the analysis period of environmental samples by 30%-50%.
Although there are some limitations, such as a high error rate (about 1%-5%) and high cost, with the progress of technology, it is expected to become mainstream in 3-5 years, which will promote the paradigm change of community research and have great potential in the fields of uncultured microorganism analysis.
Conclusion
Hierarchical research process can maximize the value of resources, while long-read may further break the boundary between them. Clarifying the differences and synergy potential between them can provide the basis for selecting precise technologies for different research objectives and promote the development of biome research in a more efficient and in-depth direction.
Comparison between DNA metabarcoding and metagenomics
| Comparison Aspect | DNA Metabarcoding | DNA Metagenomics |
|---|---|---|
| Definition | Targets specific barcode regions (e.g., 16S rRNA for bacteria) in mixed samples to identify taxa | Sequences all DNA in a sample, providing both taxonomic and functional gene information of the entire community |
| Sample Requirement | Can handle low - quality or degraded DNA, suitable for environmental samples like soil, water, and gut contents | Requires higher - quality and quantity of DNA, samples should be free of significant inhibitors |
| Workflow | Involves DNA extraction, PCR amplification of barcode regions, and high - throughput sequencing | Includes DNA extraction, library preparation without PCR (ideally), and high - throughput sequencing |
| Bioinformatics Analysis | Focuses on clustering sequences into OTUs or ASVs and taxonomic assignment | Entails sequence assembly, gene prediction, functional annotation, and taxonomic profiling at a more comprehensive level |
| Limitations | Subject to PCR biases, which can distort taxon abundances, limited to targeted barcode regions, highly dependent on reference databases | High cost, high computational demands; challenges in assembling genomes, especially for low - abundance organisms; functional predictions may not reflect actual in - situ functions |
From the five dimensions of technology selection, inherent limitations, quantitative accuracy, collaborative application, and future trends, DNA metabarcoding and metagenomics have their own adaptation scenarios:
- Choose metabarcoding if: Your priority is cost-effective, high-throughput taxonomic profiling (e.g., biodiversity surveys, biomonitoring of soil/water), or working with degraded DNA (e.g., eDNA, fossil samples).
- Choose metagenomics if: Your research demands functional insights (e.g., antibiotic resistance gene mining, metabolic pathway analysis), genome reconstruction (e.g., MAGs from uncultured microbes), or studying taxa without universal barcodes (e.g., viruses).
References
- Forbes JD, Knox NC, Peterson CL, Reimer AR. "Highlighting Clinical Metagenomics for Enhanced Diagnostic Decision-making: A Step Towards Wider Implementation." Comput Struct Biotechnol J. 2018 16: 108-120.
- Vuong P, Griffiths AP, Barbour E, Kaur P. "The buzz about honey-based biosurveys." NPJ Biodivers. 2024 3(1): 8.
- Zavadska D, Henry N, Auladell A, Berney C, Richter DJ. "Diverse patterns of correspondence between protist metabarcodes and protist metagenome-assembled genomes." PLoS One. 2024 19(6): e0303697.
- Brennan GL. "Sequencing our way to more accurate community abundance." Mol Ecol Resour. 2023 23(1): 13-15.