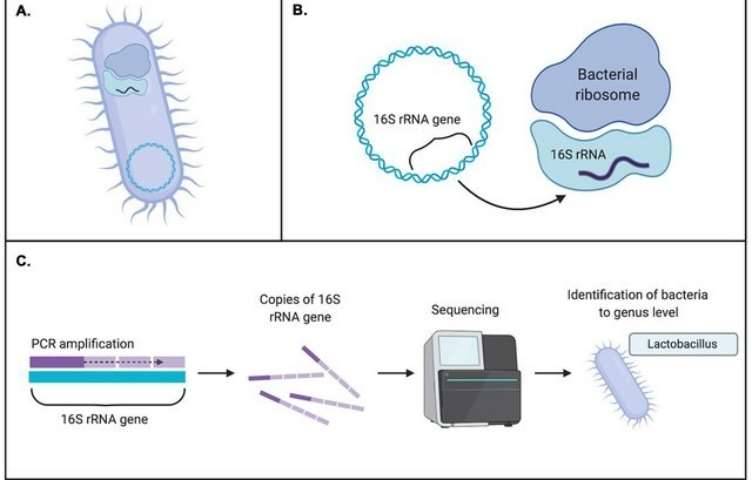
Why is Host DNA Removal Important in Metagenomic Sequencing?
In the practice of metagenomic sequencing, clinical samples, such as tissues and body fluids, often contain substantial amounts of host genomic material and inhibitory components, such as polysaccharides, bile salts, and lipid complexes. Among these, host genetic contamination represents a core challenge:
- Disparity in Gene Content: The genome size of a single human cell is approximately 3 Gb, whereas the genome of a viral particle is only 30 kb, representing a five-order magnitude difference.
- Data Dilution Effect: More than 99% of sequences in metagenomic data originate from the host, thereby completely obscuring signals from pathogenic microorganisms.
- Resource Waste: In samples with high host content (e.g., alveolar lavage fluid), over 90% of sequencing resources are ineffectively consumed.
Therefore, the establishment of effective systems for the removal of host genetic material is a critical prerequisite for metagenomic research.
Host DNA Purification in Metagenomic Sequencing
Applicable Sample Types
| Biological Matrix | Typical Scenarios |
|---|---|
| Plant and Animal Tissues | Plant roots/leaves, animal intestinal/skin/lung tissues |
| Clinical Samples | Sputum, alveolar lavage fluid, urine, oral swabs |
| Specialized Research | Gut microbiome, host-pathogen interaction studies |
Benefits of Purification
- Increased Sensitivity: Sensitivity of microbial detection increases by 1-2 orders of magnitude.
- Improved Data Validity: The proportion of target sequences increases from less than 1% to 10%-50%.
Typical Applications of Host Purification
- Biological Matrix Types: Samples from plants, animals, and insects.
- Specialized Sample Categories: Plant leaves, root samples; animal feces and various tissue samples (e.g., intestinal, skin, lung, blood).
- Body Fluid Samples: Urine, saliva, and oral swabs.
- Key Research Areas: Gut microbiome research.
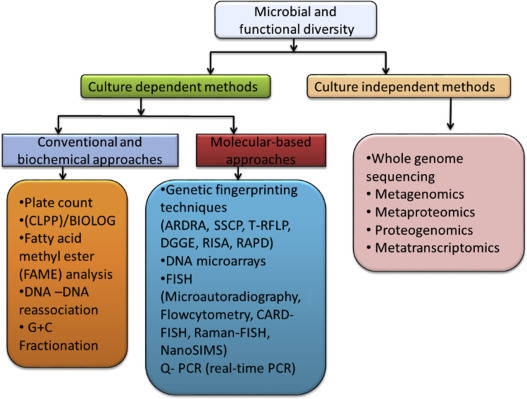
How to Remove Host DNA
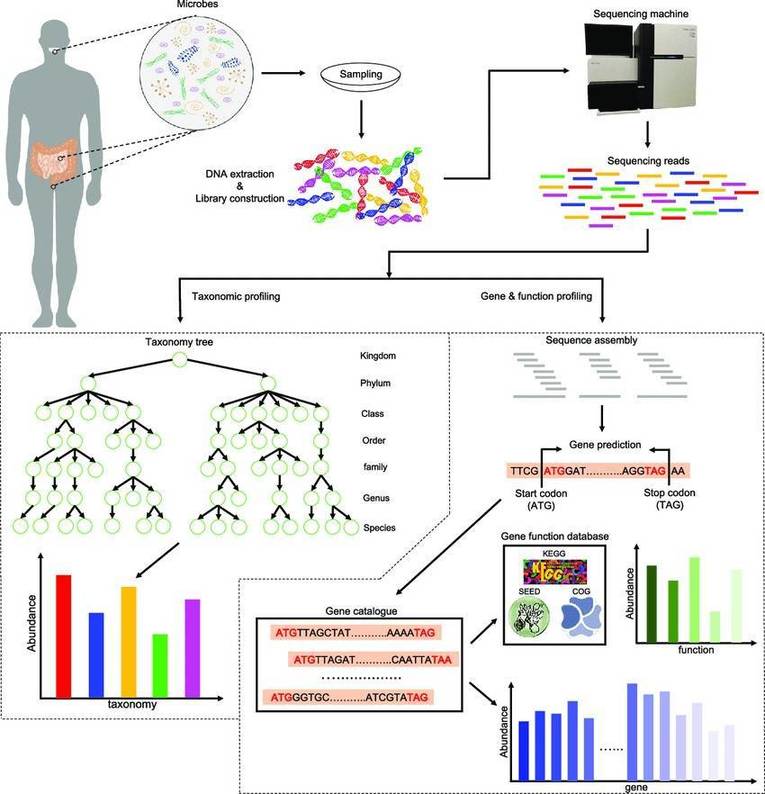
During metagenomic analysis, some samples often contain host genomes and inhibitory factors such as complex polysaccharides, bile salts, lipids, and uric acid. These elements can interfere with the sequencing of target microbial sequences. Host contamination is particularly challenging to address in certain sample types such as swabs, oral samples, and respiratory samples. The disparity in genomic size between host cells and microbial cells exacerbates the issue. For example, a human cell contains 3 Gb of genomic data, while a viral cell may contain only 30 kb, a difference of up to 100,000-fold. As a result, the vast majority of sequencing data originates from the host genome, making it difficult to detect the target microbial sequences and thereby hindering subsequent data analysis.
Thus, reducing host contamination is essential in such metagenomic samples. The common methods for host material removal are outlined below:
Methods for Host DNA Removal
1. Physical Separation Methods: Based on the physical properties of microbes and hosts.
- Centrifugal Separation: The density differences between host cells (eukaryotic cells) and bacteria/viruses can be exploited for gradient centrifugation (e.g., differential centrifugation to separate white blood cells from bacteria).
- Filtration: Filters with pore sizes ranging from 0.22 to 5 μm can trap host cells while releasing microbial DNA (suitable for enriching viruses or small bacteria).
- Limitations: This method cannot remove intracellular host DNA, such as free DNA released from lysed host cells in tissue samples.
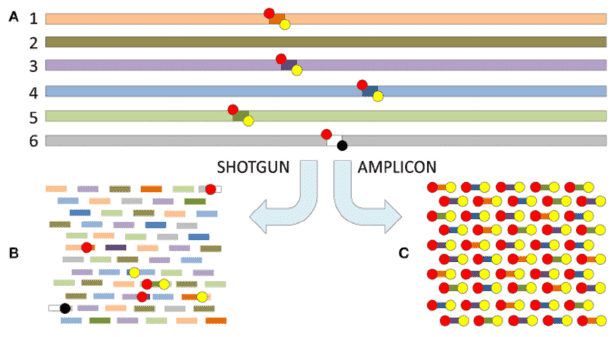
2. Targeted Amplification: Selective enrichment of microbial genomes.
- PCR Amplification: Primers are designed to target conserved microbial genes (e.g., 16S rRNA, viral capsid proteins) for specific amplification of target sequences.
- Multiple Displacement Amplification (MDA): Random primers are used to amplify low-abundance microbial DNA, suitable for ultra-low biomass samples (e.g., cerebrospinal fluid).
- Risks: Primer biases can lead to amplification discrepancies, affecting species abundance quantification.
3. Host Genome Digestion: Enzymatic and chemical cleavage methods.
- Selective Enzyme Digestion: DNase I is used to degrade free DNA (preferentially digesting host DNA fragments), combined with microbial cell wall protection strategies (e.g., bacterial fixation before lysis).
- Methylation-Sensitive Cleavage: Exploits the high methylation characteristics of host DNA (e.g., CpG islands in the human genome) to selectively cut with methylation-sensitive restriction enzymes.
- Chemical Reagents: Saponin can disrupt host cell membranes to release microbial DNA, followed by proteinase K digestion of host proteins.
4. Bioinformatics Filtering: The final defense in data cleaning.
Common Bioinformatics Tools for Host Removal
Bowtie2: A highly efficient alignment tool for mapping sequencing data (e.g., FASTQ reads) against a host reference genome.
BWA (Burrows-Wheeler Aligner): A highly accurate alignment tool, particularly suitable for high-throughput sequencing data.
KneadData: Integrates tools such as FastQC, Trimmomatic for data filtering, and Bowtie2 for host sequence removal. It includes databases for human and mouse genomes, as well as the Silva ribosomal database.
BMTagger: A tool developed by NCBI for analyzing microbiome data (e.g., FASTA, FASTQ files, or SRA datasets). Its primary function is to detect and tag sequences that may originate from human contamination.
Challenges:
These tools rely on the availability of a complete host reference genome, and they cannot remove sequences homologous to the host genome (e.g., human endogenous retroviruses).
Services you may interested in
Resource
Impact of Host DNA Removal on Metagenomic Analysis: Evidence and Case Studies
Removal of Host DNA Increases the Number of Microbial Reads
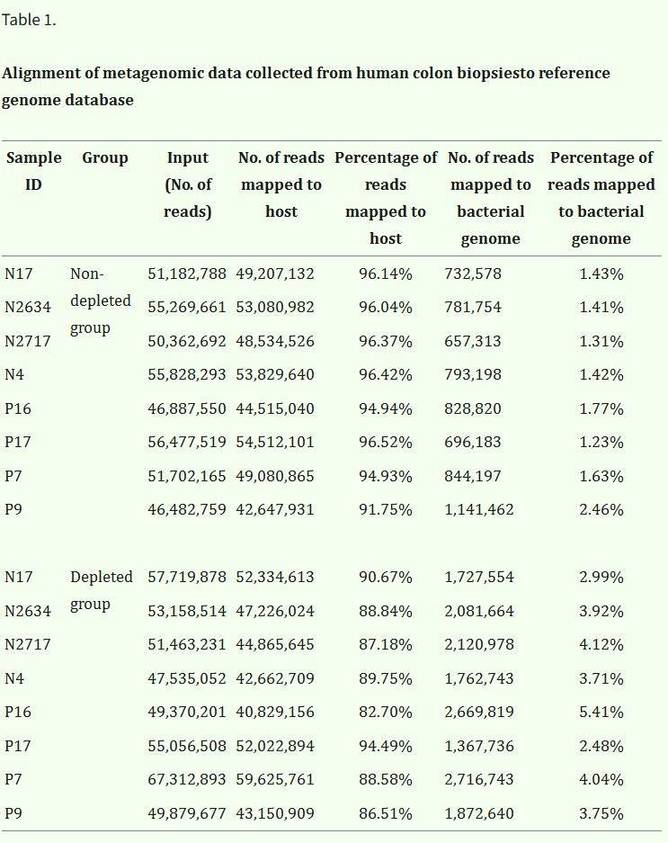
A study using samples from 8 human and 19 mouse colon biopsy samples (Table 1) generated an average of 55.37 ± 7.23 million reads per sample. In both human and mouse experimental groups, the number of bacterial reads increased, while host reads decreased (Figures 2A, C). Post-removal of host DNA, the number of bacterial species detected per sample also increased compared to the control group (Figures 2B, D). Furthermore, the majority (93.45% ± 0.89%) of the bacterial species detected in the control group were also identified in the experimental group, indicating that this method enhanced the sensitivity of species detection without disrupting the microbial composition of tissue samples.
Additionally, correlation analysis in both human and mouse samples showed a significant association between the reduction of host DNA and the increase in species detection in metagenomic sequencing (P < 0.05, Figure 2E).
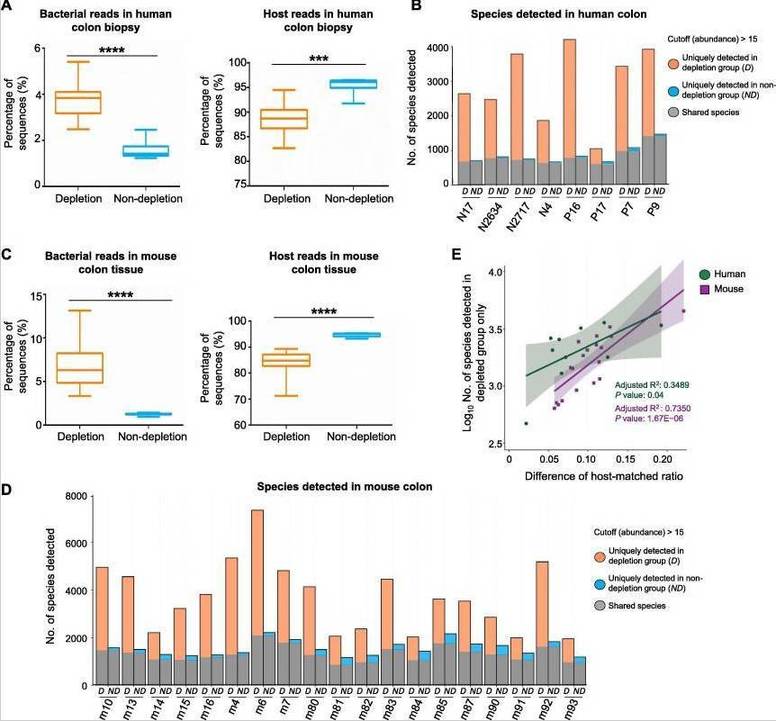
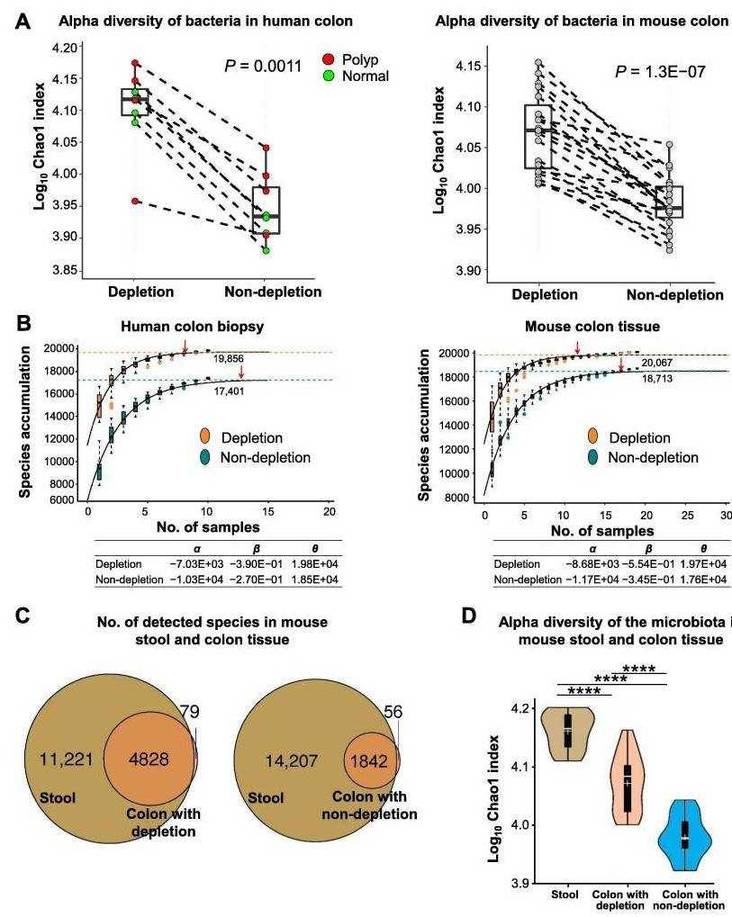
Removal of Host DNA Increases Microbial Diversity in Colon Tissue
In both human and mouse colon biopsy samples, bacterial richness significantly increased in the experimental groups (measured by the Chao1 index) following host DNA removal (Figures 3A and B). Furthermore, most bacterial species detected in mouse colon samples (98.39% in the experimental group, 97.05% in the control group) were also found in fecal samples (Figure 3C). Fecal microbiota had the highest bacterial richness, followed by the experimental group (host DNA removed) and the control group (host DNA intact) in terms of bacterial diversity (Figure 3D).
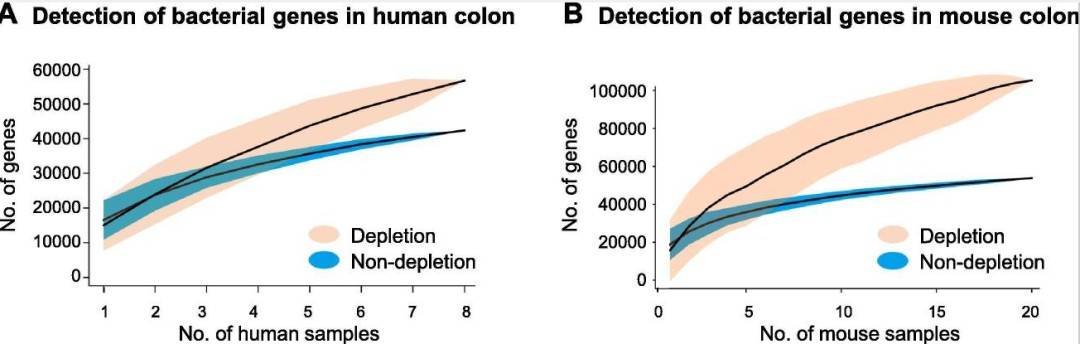
Removal of Host DNA Increases Bacterial Gene Coverage
To further evaluate the impact of host DNA removal on microbial analysis, the detection of bacterial genes in a single sample was assessed. Gene accumulation analysis showed that, after removing host DNA, the rate of bacterial gene detection increased by 33.89% in human colon biopsies (Figure 4A) and by 95.75% in mouse colon tissues compared to the control group (Figure 4B).
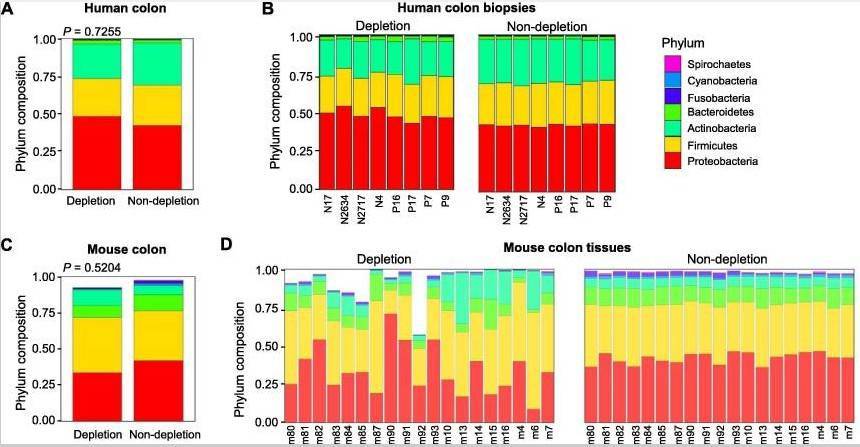
Removal of Host DNA Preserves Microbial Abundance and Increases Sequencing Depth
In both human and mouse colon tissue samples, there were no significant differences in the dominance of phyla between experimental and control groups (P < 0.05, Figure 5), suggesting that the removal of host DNA did not alter the overall structure of the microbial community. However, after host DNA removal, the abundance of certain species changed (by 0.03%). For example, several Gram-negative bacteria in human samples exhibited increased abundance after host DNA removal (Figure 6A). Further quantitative PCR (qPCR) analysis revealed that the ratio of host DNA in the experimental group and control group was similar (9.46% in qPCR vs. 10% in metagenomic sequencing). These findings suggest that the method used in this study enhanced bacterial DNA sequencing depth and revealed low-abundance bacterial species that may play significant biological roles in health maintenance or disease development.
Matching Samples to Research Objectives
| Method | Advantages | Limitations | Applicable Scenarios |
|---|---|---|---|
| Physical Separation | Low cost, rapid operation | Cannot remove intracellular host DNA | Virus enrichment, body fluid samples |
| Targeted Amplification | High specificity, strong sensitivity | Primer bias affects quantification | Low biomass, known pathogen screening |
| Host Digestion | Efficient removal of free host DNA | May damage microbial cell integrity | Tissue samples, high host content |
| Bioinformatics Filtering | No experimental manipulation, highly compatible | Dependent on reference genome, cannot remove homologous sequences | Routine samples, post-data processing |
Conclusion
Host contamination represents a major obstacle in metagenomic research. The combined use of physical separation, targeted amplification, host digestion, and bioinformatics filtering forms a four-dimensional host removal framework. Experimental (physical/chemical) methods enhance data quality before sequencing, while bioinformatics tools offer precise data cleaning post-sequencing. Different sample types (e.g., high host-content tissues vs. low biomass fluids) require customized strategies to balance sensitivity, cost, and data integrity.
References
- Cheng WY, Liu WX, Ding Y, Wang G, Shi Y, Chu ESH, Wong S, Sung JJY, Yu J. High Sensitivity of Shotgun Metagenomic Sequencing in Colon Tissue Biopsy by Host DNA Depletion. Genomics Proteomics Bioinformatics. 2023 Dec;21(6):1195-1205. DOI: 10.1016/j.gpb.2022.09.003. Epub 2022 Sep 26. PMID: 36174929; PMCID: PMC11082407.
- The Human Microbiome Project Consortium. A framework for human microbiome research. Nature 486, 215–221 (2012). https://doi.org/10.1038/nature11209
- Chiu, C.Y., Miller, S.A. Clinical metagenomics. Nat Rev Genet 20, 341–355 (2019). https://doi.org/10.1038/s41576-019-0113-7
- Heravi FS, Zakrzewski M, Vickery K, Hu H. Host DNA depletion efficiency of microbiome DNA enrichment methods in infected tissue samples. J Microbiol Methods. 2020 Mar;170:105856. DOI: 10.1016/j.mimet.2020.105856. Epub 2020 Jan 30. PMID: 32007505.
- Shi Y, Wang G, Lau HC, Yu J. Metagenomic Sequencing for Microbial DNA in Human Samples: Emerging Technological Advances. Int J Mol Sci. 2022 Feb 16;23(4):2181. DOI: 10.3390/ijms23042181. PMID: 35216302; PMCID: PMC8877284.
- Marotz, C.A., Sanders, J.G., Zuniga, C. et al. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome 6, 42 (2018). https://doi.org/10.1186/s40168-018-0426-3